
Abstract
Each month, subscribers to The Formulary Monograph Service receive 5 to 6 well-documented monographs on drugs that are newly released or are in late phase 3 trials. The monographs are targeted to Pharmacy & Therapeutics Committees. Subscribers also receive monthly 1-page summary monographs on agents that are useful for agendas and pharmacy/nursing in-services. A comprehensive target drug utilization evaluation/medication use evaluation (DUE/MUE) is also provided each month. With a subscription, the monographs are sent in print and are also available on-line. Monographs can be customized to meet the needs of a facility. Subscribers to The Formulary Monograph Service also receive access to a pharmacy bulletin board, The Formulary Information Exchange (The F.I.X.). All topics pertinent to clinical and hospital pharmacy are discussed on The F.I.X.
Through the cooperation of The Formulary, Hospital Pharmacy publishes selected reviews in this column. For more information about The Formulary Monograph Service or The F.I.X., call The Formulary at 800-322-4349. The October 2011 monograph topics are on ticagrelor, peginesatide, linaclotide, vemurafenib, and tafluprost. The DUE/MUE is on ticagrelor.
Generic Name: BOCEPREVIR
Proprietary Name: Victrelis (Merck)
Approval Rating: 1P
Therapeutic Class: Antiviral Agents; Protease Inhibitors
Similar Drugs: Telaprevir
Sound- or Look-Alike Names: Bosentan, Victoza
Indications
Boceprevir is indicated for the treatment of chronic hepatitis C genotype 1 infection in adult patients with compensated liver disease, including cirrhosis, who are treatment-naive or in whom previous therapy with interferon and ribavirin has failed. 1
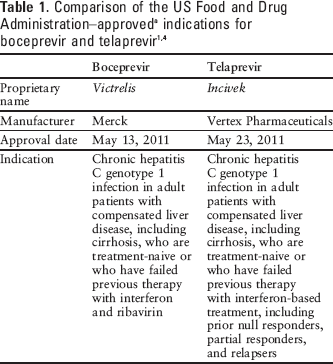
Current therapy for hepatitis C primarily consists of pegylated interferon alfa-2 and ribavirin, which can achieve a sustained virologic response (SVR) in approximately 50% of patients. Telaprevir, a similar oral hepatitis C virus (HCV) NS3/4A protease inhibitor, is also approved for use in conjunction with peginterferon alfa and ribavirin in the management of hepatitis C. 2 Table 1 compares the US Food and Drug Administration (FDA)–approved indications for boceprevir and telaprevir.
Clinical Pharmacology
Boceprevir is an oral inhibitor of HCV NS3/4A protease. 3 Maximal antiviral activity has been observed with triple-drug combinations of pegylated interferon, ribavirin, and the NS3/4 protease inhibitors (boceprevir or telaprevir).4,5
Resistance mutations at 6 positions within NS3 protease have been observed during monotherapy with boceprevir, including mutations yielding low-, medium-, and high-level resistance. The mutations also exhibited cross-resistance with telaprevir; however, there were differences in the frequency of detection and resistance levels. 6 Resistance-associated variants were also observed in approximately 20% of treatment-naive patients treated with a regimen of peginterferon alfa-2b, ribavirin, and boceprevir. 7 Naturally occurring resistance mutations, possibly associated with resistance to boceprevir, have also been observed in 0.3% to 2.8% of treatment-naive patients with genotype 1 hepatitis C infection. 8 Among patients not achieving SVR, the treatment-emergent substitutions observed most frequently among subjects infected with the HCV 1a genotype have included V36M, T54S, and R155K, and those observed most frequently among subjects infected with the HCV 1b genotype have included T54A, T54S, V55A, A156S, and I/V170A. Many treatment-emergent substitutions observed with boceprevir have been demonstrated to reduce the anti-HCV activity of other HCV NS3/4A protease inhibitors. 1
Pharmacokinetics
Following oral administration, peak concentrations (Cmax) are reached within a median of 2 hours. Steady-state peak, trough, and overall exposure increased at a less than dose-proportional extent, suggesting diminished absorption at higher doses. Overlap in exposure was observed following doses of 800 and 1,200 mg. 1
Boceprevir absorption is enhanced by administration with food. Compared with fasting, administration with food increased exposure up to 65%. Increased absorption was observed regardless of the type of meal (high fat vs low fat) and timing of the dose relative to the meal (5 minutes prior to eating, during a meal, or immediately after a meal). 1
The mean elimination half-life is 3.4 hours. 1 The majority of the dose is hepatically eliminated. Boceprevir is metabolized via ketone reduction (aldo-keto reductases AKR1C2 and AKR1C3) to inactive metabolites and to a lesser extent via cytochrome P450–mediated oxidation (CYP3A4 and CYP3A5).1,9 Approximately 3% of the dose is excreted unchanged in the urine. 1
In patients with moderate and severe hepatic impairment, exposure to the active boceprevir enantiomer was increased by 32% and 45%, respectively, compared with subjects with healthy hepatic function. 1 In patients with end-stage renal disease requiring hemodialysis, boceprevir exposure was reduced by 10% compared with subjects with healthy renal function. No dosage adjustment is necessary in patients with hepatic or renal impairment. 1 Gender, race, and age have not affected boceprevir pharmacokinetics. 1
Comparative Efficacy
Boceprevir was evaluated in studies enrolling treatment-naive patients, patients who have not responded to previous therapy with peginterferon alfa-2 plus ribavirin, and patients who relapsed following previous therapy.3,10-12 Long-term follow-up is being conducted among patients achieving SVR. 10
Boceprevir was assessed in conjunction with peginterferon alfa-2b and ribavirin in treatment-naive patients in a randomized, open-label study (SPRINT-1 [Serine Protease Inhibitor Therapy-1]) enrolling 595 patients with genotype 1 HCV infection. The genotype was 1a in 67% of patients and 1b in 32% of patients. Patients received peginterferon alfa-2b 1.5 mcg/kg plus ribavirin 800 to 1,400 mg daily for 48 weeks (104 patients); peginterferon alfa-2b plus ribavirin daily for 4 weeks, followed by peginterferon alfa-2b, ribavirin, and boceprevir 800 mg 3 times daily for 24 weeks (103 patients) or 44 weeks (103 patients); or peginterferon alfa-2b, ribavirin, and boceprevir 800 mg 3 times daily for 28 weeks (107 patients) or 48 weeks (103 patients). The 4-week lead-in with peginterferon and ribavirin was included to assess whether reaching steady state with these agents before the addition of boceprevir would lessen the emergence of drug-resistant mutations by reducing viral levels. Subsequently, 75 patients were randomly assigned to receive the triple combination for 48 weeks (16 patients) or low-dose ribavirin 400 to 1,000 mg plus peginterferon alfa-2b and ribavirin 3 times daily for 48 weeks (59 patients). The primary endpoint was SVR 24 weeks after discontinuation of treatment. Results are summarized in Table 2. All 4 boceprevir groups had higher response rates than the control group. The low-dose ribavirin regimen was less effective. Too few patients were included to determine whether there was a difference in response between those with cirrhosis and those without, or between those with baseline HCV RNA less than 600,000 units/mL compared with those with higher viral loads. Patients achieving a less than 1.5 log10 reduction in viral level after the 4-week lead-in benefited most from a treatment duration of 48 weeks, whereas those with a greater than 1.5 log10 reduction showed a similar response rate with 28 or 48 weeks of therapy. Among 109 patients who developed resistance-associated variants during treatment, 107 did not respond, relapsed, or experienced viral breakthrough; only 2 achieved SVR. Patients receiving the 4-week lead-in had the lowest frequency of resistance-associated variants.3,7
SPRINT-1 study results in treatment-naive patients with genotype 1 HCV infection 3
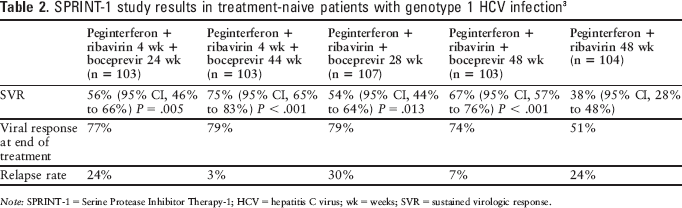
Note: SPRINT-1 = Serine Protease Inhibitor Therapy-1; HCV = hepatitis C virus; wk = weeks; SVR = sustained virologic response.
Boceprevir combined with peginterferon alfa-2b and ribavirin was also assessed in treatment-naive patients with genotype 1 HCV infection in a randomized, double-blind study enrolling 1,097 patients (SPRINT-2). The mean age was 49 years, 60% of subjects were men, and HCV RNA exceeded 400,000 units/mL in 92% of patients. Patients received a regimen that included a 4-week lead-in with peginterferon alfa-2b and ribavirin, which was followed by peginterferon and ribavirin plus placebo for 44 weeks, response-guided therapy with peginterferon and ribavirin plus boceprevir for 24 weeks, and an additional 20 weeks of peginterferon and ribavirin if HCV RNA was detectable during weeks 8 through 24, or peginterferon and ribavirin plus boceprevir for 44 weeks if HCV RNA was not detectable. Peginterferon alfa-2b was dosed weekly at 1.5 mcg/kg through subcutaneous injection. Ribavirin dosing was weight-based with a dosage of 600 to 1,400 mg/day orally divided twice daily. The boceprevir dosage was 800 mg orally 3 times daily taken with food. Therapy was discontinued at 24 weeks in patients with detectable HCV RNA. The primary efficacy endpoint was SVR 24 weeks after discontinuation of therapy. Results were analyzed separately in non-Black patients (938 patients) and Black patients (159 patients). SVR rates were consistently higher in groups treated with boceprevir (see Table 3). There was not a significant difference in response rates between those treated with the 44-week fixed regimen and those treated with response-guided therapy. Among patients with persistently undetectable HCV RNA during weeks 8 through 24, SVR was achieved in 97% (143/147) of those receiving response-guided therapy and 96% (137/142) of those receiving the fixed 44-week regimen. 11
SPRINT-2 study results for treatment-naive patients with HCV genotype 1 11

Note: SPRINT-2 = Serine Protease Inhibitor Therapy-2; HCV = hepatitis C virus; wk = weeks; SVR = sustained virologic response.
Boceprevir was also evaluated in a randomized, double-blind, placebo-controlled study enrolling 403 patients with genotype 1 hepatitis C infection who previously did not respond to, or relapsed following, treatment with peginterferon and ribavirin. The patient population was 67% men, 12% Black, and 12% with cirrhosis. Patients were randomized to receive peginterferon alfa-2b plus ribavirin for 48 weeks; peginterferon alfa-2b plus ribavirin for a 4-week lead-in followed by response-guided therapy with peginterferon alfa-2b plus ribavirin and boceprevir for 32 to 44 weeks; or peginterferon alfa-2b plus ribavirin 4-week lead-in, followed by peginterferon alfa-2b plus ribavirin and boceprevir for 44 weeks. Peginterferon alfa-2b was dosed weekly at 1.5 mcg/kg through subcutaneous injection. Ribavirin was weight-based with a dosage of 600 to 1,400 mg/day orally divided twice daily. The boceprevir dosage was 800 mg orally 3 times daily taken with food. Therapy was discontinued at week 12 if HCV RNA remained detectable. The primary endpoint was SVR at 24 weeks after discontinuation of treatment. Results are summarized in Table 4. High response rates were observed in both boceprevir groups and in previous nonresponders and patients who had relapsed after therapy with peginterferon and ribavirin. Responses were consistent across examined subgroups, including race, viral load, gender, age, genotype 1 subtype, and ALT at baseline. 12 In a combined analysis of results from SPRINT-2 and RESPOND-2 (Retreatment with HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2), higher SVR rates and lower rates of resistance-associated variants were observed in patients with the 1b genotype compared with the 1a subtype; however, SVR rates were consistently higher with boceprevir treatment in both subtypes. 13
HCV RESPOND-2 study results for patients with HCV genotype 1 who were previously unresponsive to or relapsed after treatment with peginterferon plus ribavirin therapy 12

Note: HCV = hepatitis C virus; RESPOND-2 = Retreatment with HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2; wk = weeks; SVR = sustained virologic response.
An ongoing study is enrolling subjects treated with the control regimen and not achieving SVR. These subjects are eligible to receive boceprevir plus peginterferon alfa-2b and ribavirin for up to 44 weeks. 14
Contraindications, Warnings, and Precautions
Contraindications
Boceprevir is contraindicated for use in conjunction with potent CYP3A4/5 inducers; such agents may reduce boceprevir plasma concentrations, resulting in reduced efficacy. These include carbamazepine, phenobarbital, phenytoin, rifampin, and St. John's wort. 1
Boceprevir use is also contraindicated in conjunction with CYP3A4/5 substrates for which elevated plasma concentrations are associated with serious and/or life-threatening events. These include alfuzosin, dihydroergotamine, ergonovine, ergotamine, methylergonovine, cisapride, lovastatin, simvastatin, drospirenone, pimozide, triazolam, oral midazolam, and sildenafil and tadalafil when used in the treatment of pulmonary arterial hypertension. 1
All of the contraindications associated with ribavirin and peginterferon alfa must also be followed, including use in pregnant women or men whose female partners are pregnant, and patients with hemoglobinopathies, autoimmune hepatitis, hepatic decompensation, creatinine clearance less than 50 mL/min, and hypersensitivity to any of the product components. 15 Table 5 compares the contraindications for boceprevir and telaprevir.
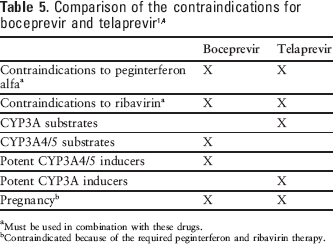
Must be used in combination with these drugs.
Contraindicated because of the required peginterferon and ribavirin therapy.
Warnings and Precautions
The addition of boceprevir to therapy with peginterferon alfa and ribavirin is associated with additional reduction in hemoglobin concentrations compared with therapy with peginterferon alfa and ribavirin alone. 1
Use of boceprevir in conjunction with peginterferon alfa and ribavirin may result in worsening of neutropenia associated with the use of peginterferon alfa and ribavirin alone. 1
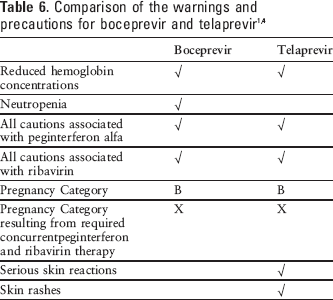
All cautions associated with peginterferon alfa and ribavirin must also be considered. 15 The warnings and precautions associated with boceprevir and telaprevir are compared in Table 6.
Boceprevir has not been studied in patients with decompensated cirrhosis or in patients who have had an organ transplant. 1
The safety and efficacy of boceprevir have not been established in patients with coinfection with HIV or hepatitis B. 1
Boceprevir is in Pregnancy Category B; however, because it must be administered in conjunction with peginterferon alfa and ribavirin, the regimen is classified in Pregnancy Category X. Ribavirin may cause birth defects and fetal death; therefore, its use is contraindicated in patients who are pregnant and in male patients with pregnant partners. Pregnancy should be avoided during therapy. Patients must have a negative pregnancy test prior to therapy, use 2 or more forms of contraception during therapy and for 6 months after treatment has concluded, and have monthly pregnancy tests. 1
Adverse Reactions
Common adverse effects observed in studies assessing boceprevir in conjunction with peginterferon alfa-2b and ribavirin included fatigue, anemia, nausea, headache, and dysgeusia.1,3,11,12 Additional adverse events reported in studies assessing boceprevir in conjunction with peginterferon alfa-2b and ribavirin are summarized in Table 7. 1
Common adverse events observed in SPRINT-1, SPRINT-2, and RESPOND-2 studies 1
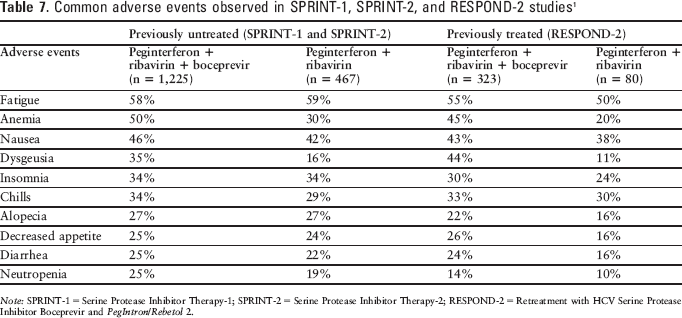
Note: SPRINT-1 = Serine Protease Inhibitor Therapy-1; SPRINT-2 = Serine Protease Inhibitor Therapy-2; RESPOND-2 = Retreatment with HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2.
Drug Interactions
Boceprevir is contraindicated for use in conjunction with potent CYP3A4/5 inducers because such agents may reduce boceprevir plasma concentrations, resulting in reduced efficacy. 1 It also should not be used in conjunction with efavirenz because this combination also results in reduced concentration of boceprevir. 1 In addition to potent CYP3A4/5 inducers, coadministration with rifabutin, dexamethasone, and ritonavir may also reduce boceprevir concentrations. Boceprevir should be used with caution, if at all, with agents known to induce CYP3A4/5. 1
Boceprevir is a strong CYP3A4/5 inhibitor. Boceprevir use is also contraindicated in conjunction with CYP3A4/5 substrates for which elevated plasma concentrations are associated with serious and/or life-threatening events. 1 Labeling also advises against the concomitant use of inhaled budesonide, fluticasone, or salmeterol. Caution is also advised and dosage adjustments are recommended if boceprevir is coadministered with amiodarone, bepridil, flecainide, propafenone, quinidine, digoxin, warfarin, trazodone, desipramine, ketoconazole, itraconazole, posaconazole, voriconazole, colchicine, clarithromycin, felodipine, nifedipine, nicardipine, bosentan, atorvastatin, cyclosporine, sirolimus, tacrolimus, methadone, buprenorphine, oral contraceptives, sildenafil, tadalafil, vardenafil, alprazolam, and intravenous midazolam. 1
The manufacturer has committed to conduct postmarketing studies to assess for potential drug interactions between boceprevir and oral contraceptives, methadone, P-glycoprotein substrates, selective serotonin reuptake inhibitors, tacrolimus, cyclosporine, and prednisone. 16
Recommended Monitoring
Complete blood cell counts (with white blood cell differential counts) should be obtained before treatment, at treatment weeks 4, 8, and 12, and as clinically necessary. HCV RNA should be monitored at treatment weeks 4, 8, 12, and 24; at the end of treatment; and during follow-up. 1 All monitoring recommended for peginterferon alfa and ribavirin must be observed, including hematologic and blood chemistry testing, pregnancy testing, and monitoring for worsening of depression or other psychiatric symptoms. 15 Table 8 compares the recommended monitoring for boceprevir and telaprevir.

Note: HCV = hepatitis C virus; wk = weeks.
Dosing
The recommended dosage is boceprevir 800 mg orally 3 times daily (every 7 to 9 hours) taken with food (a meal or light snack). Boceprevir must be coadministered with peginterferon alfa and ribavirin. 1 No dosage adjustments are necessary in patients with renal or hepatic impairment. 1 Dosage reductions of boceprevir are not recommended for any reason. 1
For all patients, therapy should be initiated with peginterferon alfa and ribavirin for 4 weeks prior to starting boceprevir therapy (treatment weeks 1 through 4). Boceprevir 800 mg 3 times daily should be added after 4 weeks of treatment. 1 A 4-week lead-in phase with peginterferon alfa and ribavirin prior to initiation of boceprevir has been associated with higher rates of SVR and fewer occurrences of resistance-associated variants.3,7,11,12 The recommended duration of boceprevir therapy ranges from 24 to 44 weeks (see Table 9), depending on previous treatment status, HCV RNA at weeks 8 and 24, and presence of cirrhosis. 1 Treatment should be discontinued in patients with HCV RNA levels of 100 units/mL or more at treatment week 12 or confirmed detectable HCV RNA at treatment week 24. 1
Recommended duration of therapy for boceprevir 1
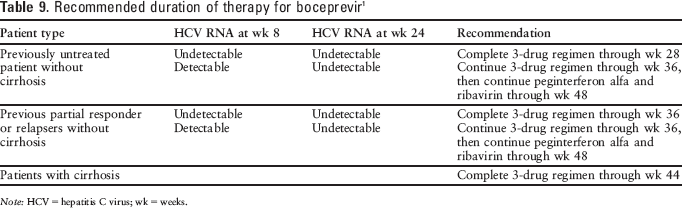
Note: HCV = hepatitis C virus; wk = weeks.
Product Availability and Storage
Boceprevir received US FDA approval on May 13, 2011. It is available as a 200 mg capsule packaged in cartons of 28 bottles, each containing 12 capsules (a 1-day supply). Boceprevir should be stored refrigerated between 2°C and 8°C (36°F and 46°F) until dispensed. The refrigerated capsules are stable through the expiration date printed on the label. For patient use, boceprevir can also be stored at room temperature, up to 25°C (77°F) for 3 months. 1
Risk Evaluation and Mitigation Strategy (Rems)
No REMS is required for boceprevir.
Conclusion
The addition of boceprevir to a standard regimen of peginterferon alfa and ribavirin has exhibited enhanced SVR without a substantial increase in toxicity in patients with genotype 1 hepatitis C who are treatment-naive or in whom previous treatment has failed. Although boceprevir has been relatively well tolerated, careful assessment for potential drug interactions is necessary when considering boceprevir therapy. Boceprevir and telaprevir are both likely to be widely incorporated into treatment regimens for hepatitis C. Comparative data with telaprevir will be valuable, particularly to help discern any differences in the adverse effect profiles of these 2 drugs. At this time, no head-to-head studies have been conducted, and the individual studies with boceprevir and telaprevir have used the agents in conjunction with different pegylated interferons, with different ribavirin doses, and for differing durations of therapy, making direct comparisons difficult.
Generic Name: TELAPREVIR
Proprietary Name: Incivek (Vertex)
Approval Rating: 1P
Therapeutic Class: Antiviral Agents; Protease Inhibitors
Similar Drugs: Boceprevir
Sound- or Look-Alike
Names: Oseltamivir, Peramivir, Tenofovir, Tipranavir
Indications
Telaprevir is approved for the treatment of chronic hepatitis C genotype 1 infections in combination with peginterferon alfa and ribavirin in adult patients with compensated liver disease, including cirrhosis, who are treatment-naive or have not responded to previous therapy with interferon-based treatment, including prior null responders, partial responders, and relapsers.4,17 However, a high proportion of the previous null responders, especially those with cirrhosis, do not achieve a sustained viral response (SVR) and have telaprevir resistance–associated substitutions emerge while receiving telaprevir therapy; efficacy has not been established in patients with a history of failed response to telaprevir or other hepatitis C virus (HCV) NS3/4A protease inhibitors. 4
Current standard of care for hepatitis C primarily consists of pegylated interferon alfa-2 and ribavirin, which achieve SVR in approximately 50% of patients. The achievement of an SVR after completing therapy is a surrogate marker for a therapy-induced cure, which would be associated with a decreased risk of developing cirrhosis or other complications of liver disease, decreased rates of liver cancer (eg, hepatocellular carcinoma), and decreased mortality.2,18-20 Boceprevir, a similar oral HCV NS3/4A protease inhibitor, is also approved for the treatment of chronic hepatitis C genotype 1 infection in adult patients with compensated liver disease, including cirrhosis, who are treatment-naive or have not responded to previous therapy with interferon and ribavirin. 1 Neither of these drugs are recommended as monotherapy and should only be used in combination with peginterferon alfa and ribavirin.4,1,20-24 Table 1 compares the US FDA-approved indications for boceprevir and telaprevir.
Clinical Pharmacology
Telaprevir is an oral inhibitor of HCV NS3/4A protease. Inhibition of NS3/4A protease may block viral replication and restore control of HCV infection by interferon alpha–mediated pathways. 2
With a 14-day regimen, telaprevir monotherapy reduced HCV RNA by 3.99 log10 copies/mL from baseline, and a combined regimen with peginterferon alfa-2a reduced HCV RNA by 5.49 log10 copies/mL compared with a reduction of 1.09 log10 copies/mL with peginterferon alfa-2a alone. 25 With monotherapy, a reduction of 4.77 log10 units/mL was observed within 3 to 7 days; however, viral breakthrough was observed in some patients. 26 A combined regimen of telaprevir with pegylated interferon alfa or other direct antiviral drugs is necessary to avoid rapid development of telaprevir resistance.26,27
Resistance or decreased susceptibility has been reported in cell culture assays, animal models, and clinical studies. The decreased susceptibility to telaprevir was a result of variants V36A/M, V36A/M/L, V36C, T54A/S, R155K/T, A1568, A1568/T, R155T+D168N, V36A+T54A, A156V/T, V36M/A+R155K/T, and T54S/A+A1568/T. Susceptibility with these variants decreased from 3-fold to up to 62-fold. Various types of these variants were found in some patients who did not achieve SVR. On-treatment virologic failure generally occurred in patients with genotype 1a more than those with genotype 1b and more frequently in those who were prior null responders.4,23,28-31
Cross-resistance may occur with boceprevir and other HCV NS3/4A protease inhibitors, especially in patients who did not achieve SVR and had treatment-emergent NS3 amino acid substitutions at positions V36, T54, R155, A156, or D168. However, the impact of prior exposure to telaprevir or treatment failure with telaprevir-based therapy on boceprevir or other HCV NS3/4A protease inhibitors has not been studied, nor has the impact of previous treatment with a NS3/4A protease inhibitor on the efficacy of telaprevir. 4
A strong predictor of response to peginterferon alfa and ribavirin therapy is the presence of the genetic variant near the gene-encoding interferon-lambda-3 (IL28B rs12979860, a C to T change). Those with the C/C genotype tended to have a better SVR, whereas those with the C/T or T/T genotypes had a trend to lower SVR rates.4,32-34
Pharmacokinetics
Telaprevir is orally bioavailable. 2 The majority of it is absorbed from the small intestine. Peak plasma concentrations occur within 5 hours of oral administration. Administration with a high-fat meal increases the area under the curve by 237%. In clinical trials, patients took the telaprevir dose within 30 minutes of a meal or snack that contained approximately 20 g of fat. 4
Protein binding to plasma protein varies from 59% to 76%. Binding to the plasma alpha-1 acid glycoprotein and albumin was inversely related to plasma concentration; decreased plasma protein binding occurred with increased plasma concentrations. The average apparent volume of distribution was 252 L. 4
Telaprevir is extensively metabolized by the liver. 4 Because of its high lipophilicity, first-pass uptake concentrates the drug in the liver. Average liver concentrations were more than 35 times those in plasma in an animal model. 2 The metabolic pathways include hydrolysis, oxidation, and reduction. Multiple metabolites can be found in the feces, plasma, and urine. Cytochrome P450 is also involved with the metabolism of telaprevir and the CYP3A4 isoform responsible for this metabolic pathway. 4 The apparent total clearance is 32.4 L/h, the mean elimination half-life after single-dose administration is 4 to 4.7 hours, and the steady-state half-life is 9 to 11 hours. 4
Gender, race, age, and renal impairment have little to no impact on the pharmacokinetics of telaprevir. Changes in pediatric patients have not been evaluated.
Telaprevir is not recommended for patients with moderate or severe hepatic impairment because the appropriate dose has not been determined. Steady-state exposure to telaprevir is reduced by 15% in HCV-negative patients with mild hepatic impairment (Child-Pugh class A) and 46% in HCV-negative patients with moderate hepatic impairment (Child-Pugh class B). 4
Comparative Efficacy
All patients enrolled in the pivotal trials had genotype 1 chronic hepatitis C. These patients were treatment-naive, had failed to respond to treatment, or had relapsed following previous therapy. They all had compensated liver disease, detectable HCV RNA, and liver histopathology consistent with chronic hepatitis C. The dosage of telaprevir was 750 mg every 8 hours in combination with peginterferon alfa 180 mcg/week and ribavirin 1,000 or 1,200 mg/day, depending on the patient's body weight. SVR was defined as an HCV RNA level of less than 25 units/mL at 24 weeks after the planned end of treatment. 4
The ADVANCE (A New Direction in HCV Care: a Study of Treatment-Naive Hepatitis C Patients With Telaprevir) study was a phase 3, randomized, double-blind, placebo-controlled trial comparing 2 telaprevir regimens in conjunction with peginterferon alfa-2a and ribavirin with a regimen of peginterferon alfa-2a plus ribavirin alone. The study enrolled 1,095 treatment-naive patients with genotype 1 hepatitis C; 77% had HCV RNA greater than 800,000 units/mL, 58% had genotype 1a HCV, 21% had bridging fibrosis or compensated cirrhosis, 58% were men, 9% were Black, and 11% were Hispanic. Patients assigned to a telaprevir regimen received telaprevir 750 mg every 8 hours for 8 or 12 weeks in combination with standard doses of peginterferon alfa-2a (180 mcg once weekly) and ribavirin (1,000 or 1,200 mg weight-based oral daily dose) followed by therapy with peginterferon alfa-2a and ribavirin alone. In patients achieving undetectable virus levels at weeks 4 and 12 after starting therapy (extended rapid viral response), the total length of therapy was 24 weeks; for all others, therapy was continued for a total of 48 weeks. The primary study endpoint was SVR, defined as undetectable virus in the blood 24 weeks after all treatment ended. SVR was achieved in 69% of patients treated with telaprevir for 8 weeks (P < .001) and 75% of patients treated with telaprevir for 12 weeks (P < .001) compared with 46% of patients treated only with peginterferon alfa-2a and ribavirin. Viral relapse, defined as the proportion of patients who achieved undetectable HCV RNA at the completion of all treatment but relapsed during posttreatment follow-up, occurred in 8.6% of patients in the 12-week arm and in 9.5% of patients in the 8-week arm compared with 28% of patients in the control group. Virologic failure after week 12 was higher in the 8-week arm (10.2%) compared with the 12-week arm (5%) and was primarily associated with wild-type and lower level telaprevir-resistant variants. Although noninferiority was established at endpoint, it was suggested that the extra 3 weeks of therapy may reduce subsequent virologic failures.4,24,35-37
The ILLUMINATE (Illustrating the Effects of Combination Therapy With Telaprevir) study was a phase 3, open-label study with no control arm that compared 24- and 48-week therapy for patients with genotype 1 hepatitis C who achieved undetectable virus levels at weeks 4 and 12 of treatment (extended rapid viral response) and who remained in the trial through week 20. The study enrolled 540 treatment-naive patients with genotype 1 hepatitis C. Enrolled patients were mostly men (60.2%) and White (79.1%; Black, 13.5%), with a median HCV RNA of log10 6.5 units/mL; 11.3% of the patients had cirrhosis. All patients received telaprevir 750 mg every 8 hours in combination with standard doses of peginterferon alfa-2a and ribavirin for 12 weeks followed by therapy with peginterferon alfa-2a and ribavirin alone. Extended rapid viral response was achieved in 72% (389) of patients; extended rapid viral response was achieved in 65.2% (352) of patients. Of these, 322 who met criteria were randomized at week 20 to receive 24 or 48 total weeks of therapy. All others continued peginterferon alfa-2a and ribavirin therapy for a total of 48 weeks. The primary endpoint was noninferiority with respect to SVR in the 24- and 48-week treatment groups, which was met with SVR rates of 92% with the 24-week regimen and 87.5% with the 48-week regimen. Additional study results are included in Table 10.4,38-40

Note: ILLUMINATE = Illustrating the Effects of Combination Therapy With Telaprevir; wk = weeks; SVR = sustained virologic response; ITT = intent-to-treat.
A pooled analysis of the ADVANCE and ILLUMINATE studies was done to evaluate the impact of race or ethnicity on telaprevir's efficacy. The improvements in SVR rates in Black and Hispanic/Latino patients with the triple-drug regimen was better than those observed in the dual-drug regimen. 41 The triple-drug regimen was also associated with less treatment-regimen fatigue. 42
The REALIZE (Re-treatment of Patients With Telaprevir-based Regimen to Optimize Outcomes) study was a phase 3, randomized, controlled study comparing 2 different regimens of telaprevir plus peginterferon alfa-2a and ribavirin (delayed and simultaneous start of telaprevir for a total of 48 weeks of treatment) versus a 48-week regimen of only peginterferon alfa-2a plus ribavirin. The study included 662 patients with genotype 1 hepatitis C who did not achieve SVR with a prior pegylated interferon–based treatment, including difficult to treat null-responder patients (28%) and patients who had a partial response (19%) or relapse (53%) to prior therapy. Cirrhosis was present in 26% of patients and 89% had a high viral load (HCV RNA 800,000 units/mL or higher) at study entry. Two telaprevir-containing regimens (telaprevir 750 mg every 8 hours in combination with peginterferon alfa-2a and ribavirin for 12 weeks followed by therapy with peginterferon alfa-2a and ribavirin alone for 36 weeks, and 4 weeks of peginterferon alfa-2a plus ribavirin followed by 12 weeks of therapy with the combination of telaprevir with peginterferon alfa-2a and ribavirin and then another 32 weeks of therapy with peginterferon alfa-2a plus ribavirin) were compared with a regimen of 48 weeks of peginterferon alfa-2a plus ribavirin. The primary endpoint was SVR, which was achieved in 65% of patients treated with a telaprevir regimen compared with 17% in the control arm. Results were similar in the 2 telaprevir groups, demonstrating no advantage to the delayed-treatment start. Results from the 2 telaprevir arms are summarized in Table 11.4,43,44

Note: REALIZE = Re-treatment of Patients With Telaprevir-based Regimen to Optimize Outcomes; SVR = sustained virologic response; ITT = intent-to-treat.
P < .001 vs control.
P < .001.
Telaprevir was also assessed in a phase 2, randomized, double-blind, placebo-controlled study including 250 treatment-naive patients with genotype 1 HCV (the PROVE1 [Protease Inhibition for Viral Evaluation 1] study). Study patients were mostly men (63%) and White (77%), with a mean age of 48.1 years. Patients in the control group received peginterferon alfa-2a 180 mcg/week plus ribavirin 1,000 or 1,200 mg daily for 48 weeks plus concomitant placebo for the first 12 weeks. Patients in the telaprevir group received 1,250 mg on day 1 and 750 mg every 8 hours for 12 weeks in addition to peginterferon alfa-2a and ribavirin for 12 weeks (17 patients) or a total of 24 (79 patients) or 48 weeks (79 patients). SVR, the primary study endpoint, was achieved in 41% of patients in the control group compared with 61% of patients in the 24-week group (P = .02) and 67% of patients in the 48-week group (P = .002). Viral breakthrough occurred in 7% of telaprevir-treated patients. 45
Telaprevir was also assessed in a phase 2, randomized, partially blinded study including 334 treatment-naive patients with genotype 1 HCV (the PROVE2 study). Study patients were mostly male (approximately 60%) and White (approximately 93%), with a median age of approximately 45 years. Patients in the control group received peginterferon alfa-2a 180 mcg weekly plus ribavirin 1,000 or 1,200 mg daily for 48 weeks. Telaprevir treatment groups received 1,250 mg on day 1 followed by 750 mg every 8 hours. One group received telaprevir for 12 weeks concomitantly with peginterferon alfa-2a and ribavirin. Another group received telaprevir plus peginterferon alfa-2a and ribavirin for 12 weeks, plus an additional 12 weeks of therapy with peginterferon alfa-2a and ribavirin for a total of 24 weeks of therapy. A third telaprevir group received telaprevir for 12 weeks in conjunction with peginterferon alfa-2a for 12 weeks with no ribavirin. SVR, the primary study endpoint, was achieved in 60% of patients treated with telaprevir plus peginterferon and ribavirin for 12 weeks (P = .12 vs control), 36% of patients treated with telaprevir plus peginterferon for 12 weeks (P = .2 vs control), and 69% of patients treated with telaprevir for 12 weeks plus peginterferon plus ribavirin for a total of 24 weeks (P = .004 vs control) compared with 46% in the control group. The proportion of patients with undetectable HCV RNA at weeks 4 and 12 was higher in the telaprevir groups than in the control group (50% to 80% vs 13%; P < .001 for each regimen). SVR rates were highest in the regimens including ribavirin and with regimens extending peginterferon and ribavirin therapy beyond the telaprevir regimen. 46
A third phase 2 (PROVE3), randomized, partially blinded study assessed telaprevir in 453 patients who had not had an SVR after therapy with peginterferon alfa plus ribavirin. The majority of study patients were male (approximately 68%) and White (approximately 89%), with a median age of 50 to 53 years. Following previous therapy, approximately 57% of patients were classified as nonresponders, approximately 36% had relapsed, and approximately 7% had experienced viral breakthrough. Patients received telaprevir 1,125 mg as a loading dose followed by 750 mg every 8 hours for 12 weeks in conjunction with peginterferon and ribavirin for a total of 24 weeks (115 patients), telaprevir for 24 weeks plus peginterferon and ribavirin for 48 weeks (113 patients), telaprevir and peginterferon alfa-2a for 24 weeks (111 patients), or peginterferon plus ribavirin for 48 weeks (114 patients). SVR, the primary study endpoint, was achieved in 51% of patients receiving telaprevir plus peginterferon and ribavirin for 24 weeks (P < .001 vs control), 53% receiving telaprevir plus peginterferon and ribavirin for 48 weeks (P < .001 vs control), and 24% receiving telaprevir with peginterferon alone (P = .02 vs control) compared with 14% in the control group not receiving telaprevir.47–49
The EXTEND study was a 3-year virology follow-up study to the phase 2 clinical trials. Of 867 patients who received at least 1 dose of telaprevir in PROVE1, PROVE2, PROVE3, or Study 107 and who had baseline HCV sequences that were eligible for enrollment, 202 patients entered the study. The 123 patients who achieved SVR were observed for a median of 22 months (range, 5 to 35 months) following SVR. Those who did not achieve SVR (79 patients) were observed for a median of 25 months (range, 7 to 36 months) after the end of the previous study. Of the 123 patients who achieved SVR, 122 (99%) maintained SVR during follow-up. Among patients who did not achieve SVR after telaprevir therapy, variants associated with reduced susceptibility to telaprevir were no longer detectable in 89% of patients within the follow-up period. 50
C210 was a study of telaprevir administered as 750 mg every 8 hours as monotherapy (8 patients) or at the same dose with peginterferon alfa-2a 180 mcg/wk and ribavirin 1,000 or 1,200 mg/day (8 patients), or placebo with peginterferon plus ribavirin (8 patients) for 15 days in patients with genotype 4 HCV. All patients continued therapy with peginterferon alfa-2a and ribavirin to complete a total of 48 weeks of therapy. An increase in viral load was observed in 5 patients receiving telaprevir monotherapy compared with none receiving telaprevir in combination with peginterferon alfa-2a and ribavirin. The median log10 copies/mL HCV RNA change from baseline to day 15 was −0.77 with the sequential regimen, −4.32 with the concomitant regimen, and −1.58 with the placebo regimen. SVR was achieved in 62.5% of patients treated with the sequential regimen, 50% treated with the concomitant regimen, and 62.5% treated with the placebo regimen. Although coadministration for 15 days was associated with a greater early reduction in viral load, SVR did not differ with the addition of telaprevir to the regimen of peginterferon alfa-2a and ribavirin in patients with genotype 4 HCV. Additional studies evaluating longer use of telaprevir are necessary. 51
An additional study is also underway assessing telaprevir in combination with an HCV polymerase inhibitor (VX-222). 38
The following postmarketing studies are required 17 :
Pharmacokinetic study to determine appropriate dosing for treatment-naive pediatric patients (3 to 17 years of age)
Safety and efficacy study in combination with pegylated interferon and ribavirin in pediatric patients (3 to 17 years of age), enrolling treatment-naive patients and patients who have failed a prior course of pegylated interferon and ribavirin therapy
Susceptibility study in the HCV replicon system to determine the impact of various emergent amino acid substitutions on phenotypic susceptibility to telaprevir
Retrospective evaluation of the phase 3 data for patients with virologic failure without detected NS3/4A resistance–associated substitutions for the presence of substitutions in NS3/4A protease cleavage sites
Pharmacokinetic evaluation in adults with end-stage renal disease on intermittent hemodialysis
Safety and response study in treatment-naive and -experienced patients with cirrhosis and a comparison of the results with those from patients without cirrhosis
Safety and response comparison of Black patients with non-Black patients
Safety and efficacy evaluation in treatment-naive and treatment-experienced HIV/HVC coinfected patients
Genomic assessment to determine risk factors for the development of severe rash and severe cutaneous adverse reactions
Contraindications, Warnings, and Precautions
Contraindications
Telaprevir is contraindicated for use in conjunction with drugs that are highly dependent on CYP3A for clearance and may cause serious and/or life-threatening events with elevated plasma concentrations. It is also contraindicated in patients requiring therapy with a drug that is a strong inducer of CYP3A because subtherapeutic levels of telaprevir may occur (see
Contraindicated concurrent drug therapy with telaprevir 4
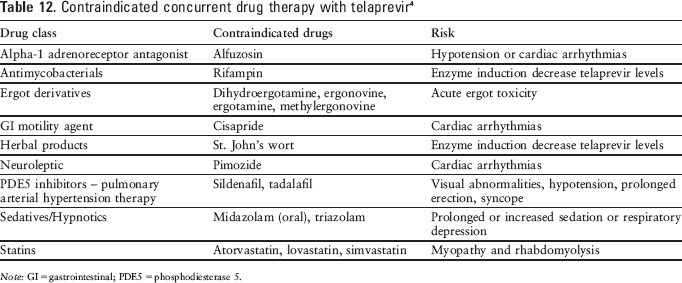
Note: GI = gastrointestinal; PDE5 = phosphodiesterase 5.
All of the contraindications associated with ribavirin and peginterferon alpha must also be followed, including use in pregnant women or men whose female partners are pregnant, and patients with hemoglobinopathies, autoimmune hepatitis, hepatic decompensation, creatinine clearance (CrCl) less than 50 mL/min, and hypersensitivity to any of the product components. 15
Warnings and Precautions
Skin reactions with telaprevir therapy, especially rash, are common. If the skin rash is mild to moderate in severity, it should be monitored for progression. If the rash gets worse, the telaprevir therapy should be discontinued. If the patient develops a serious skin reaction (eg, drug rash with eosinophilia and systemic symptoms [DRESS] syndrome, Stevens-Johnson syndrome) the telaprevir, peginterferon alfa, and ribavirin therapy should be discontinued immediately. 4 Treatment of rashes with oral antihistamines and/or topical corticosteroids may provide some symptomatic relief, but their effectiveness has not been established. The use of systemic corticosteroids is not recommended for the treatment of rash. 4
The addition of telaprevir to peginterferon alfa plus ribavirin therapy can cause an additional reduction in hemoglobin concentrations compared with therapy with peginterferon alfa and ribavirin alone. Hemoglobin of 10 g/dL or less was observed in 36% of the patients receiving triple-drug therapy versus 17% with the peginterferon alfa/ribavirin combination therapy. Hemoglobin levels less than 8.5 g/dL were observed in 14% and 5%, respectively. Hemoglobin levels should be monitored prior to the start of telaprevir and every 4 weeks throughout therapy. If the levels decrease, the dose of ribavirin should be adjusted. If the levels remain abnormal, discontinuing the telaprevir therapy is advised. 4 The risk of developing anemia appears to be higher in patients with the in-osine triphosphatase gene, especially those with the AA genotype at rs7270101 and CC genotype at rs1127354. 52 The development of anemia did not effect the efficacy observed in treatment-naive patients in the ADVANCE or ILLUMINATE studies. 53
All cautions associated with peginterferon alfa and ribavirin must also be considered. 15 Table 6 compares the warnings and precautions associated with boceprevir and telaprevir.
The safety and efficacy of telaprevir have not been established in pediatric patients. 4
The safety and efficacy of telaprevir have not been established in patients with coinfection with HIV or hepatitis B.
The safety and efficacy of telaprevir have not been established in solid organ transplant recipients. 4
Telaprevir is classified as Pregnancy Category B. However, because it must be coadministered with peginterferon alfa and ribavirin, the regimen is classified in Pregnancy Category X. Ribavirin may cause birth defects and fetal death; therefore, its use is contraindicated in patients who are pregnant and in male patients with pregnant partners. Pregnancy should be avoided during therapy. Patients must have a negative pregnancy test prior to therapy, use 2 or more forms of contraception during therapy and for 6 months after treatment has concluded, and have monthly pregnancy tests. 4
Adverse Reactions
The most common adverse events reported during telaprevir dosing were rash, pruritus, anemia, nausea, hemorrhoids, diarrhea, anorectal discomfort, dysgeusia, fatigue, and vomiting (see Table 13).4,35,36,39,43,45,46 In the PROVE studies, asthenia, nausea, pruritus, and rash occurred more frequently in the telaprevir regimens, with incidences close to 50%.45–47
Adverse reactions reported in the telaprevir clinical trials 4
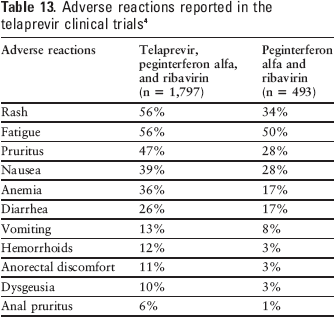
While skin reactions are common, the development of serious skin reactions (eg, DRESS syndrome, Stevens-Johnson syndrome) associated with telaprevir are uncommon. Most rashes occurred within the first 4 weeks of therapy, but can occur at any time. There was improvement of rash after drug discontinuation and the rash generally resolved within weeks.4,54
Neutropenia is listed as a warning/precaution with boceprevir, but not with telaprevir. However, decreased white blood cell counts have been observed in patients treated with telaprevir. Lymphocyte counts less than 500/mm3 occurred in 15% of patients treated with the triple-drug regimen compared with 5% of patients treated with the double-drug regimen. White blood cell counts less than 1,500/mm3 were comparable between the 2 groups (8% with triple-drug therapy and 5% with double-drug therapy). An absolute neutrophil count less than 750/mm3 occurred in 15% of patients treated with the triple-drug regimen and 12% of patients treated with the double-drug regimen. In addition, a decrease in mean platelets count occurred in 47% and 36% of patients, respectively, and less than 50,000/mm3 in 3% and 1% of patients, respectively. 4
Uric acid levels may be increased during therapy with telaprevir. Elevations in uric acid levels were observed in 73% of patients treated with the triple-drug regimen and 28% of patients treated with the double-drug regimen. The percentage of patients with uric acid levels greater than or equal to 2.6 × upper limit of normal were 4% with the triple-drug regimen and 2% with the double-drug regimen. 4
Drug Interactions
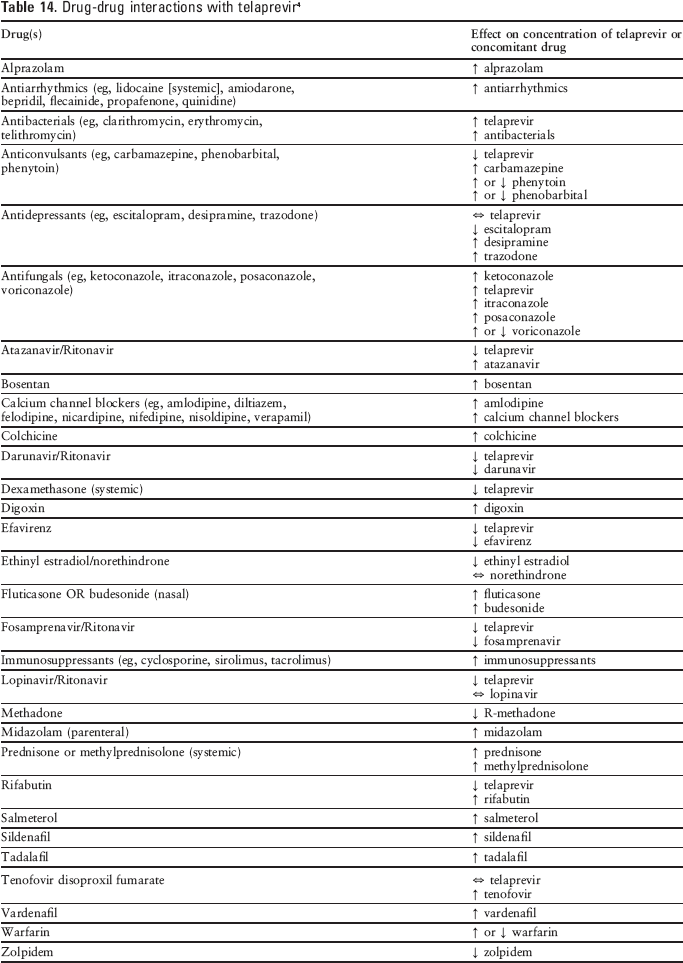
Telaprevir therapy is associated with a large number of potential drug-drug interactions because it is a substrate and inhibitor of CYP3A and P-glycoprotein (Pgp) (see Table 14). Concurrent use of low therapeutic index drugs metabolized by CYP3A and Pgp may result in elevated plasma levels of the other drug and potential toxicity, while CYP3A inducers may lower the plasma levels of telaprevir and decrease its efficacy. 1
Drug-drug interactions with telaprevir 4
Recommended Monitoring
Baseline hemoglobin assessment is recommended along with periodic assessments throughout therapy. If the patient is female and of childbearing age, she needs to have pregnancy tests prior to the start of therapy and monthly thereafter. 4 Measurement of HCV RNA levels are also recommended at weeks 4 and 12 of telaprevir therapy for all patients. 4
All monitoring recommended for peginterferon alfa and ribavirin must be observed, including hematologic and blood chemistry testing, pregnancy testing, and monitoring for worsening of depression or other psychiatric symptoms. 15 Recommended monitoring for boceprevir and telaprevir are compared in Table 8.
Dosing
The approved dosage of telaprevir is 750 mg 3 times daily (7 to 9 hours apart) with food (not low fat) in combination with a peginterferon alfa and ribavirin regimen. Triple therapy is initiated with telaprevir, peginterferon alfa, and ribavirin for 12 weeks, then peginterferon alfa and ribavirin should be continued for 12 or 36 weeks, depending on the patient's viral response and prior response status. 4
The duration of treatment of telaprevir is determined by the patient's viral response, HCV RNA levels, and prior response status. Patients who are treatment-naive or those with a prior relapse with an undetectable HCV RNA at weeks 4 and 12 should receive telaprevir for 12 weeks along with peginterferon alfa and ribavirin followed by an additional 12 weeks of peginterferon alfa plus ribavirin therapy. If the HCV RNA is detectable (1,000 units/mL or less) at weeks 4 and/or 12, telaprevir is given for 12 weeks along with the peginterferon alfa and ribavirin, followed by an additional 36 weeks of peginterferon alfa plus ribavirin therapy. If the patient has been classified as a prior partial or null responder, they should receive telaprevir plus peginterferon alfa and ribavirin for 12 weeks followed by an additional 36 weeks of peginterferon alfa plus ribavirin therapy. Treatment-naive patients with cirrhosis and an undetectable HCV RNA level at weeks 4 and 12 of telaprevir plus peginterferon alfa and ribavirin therapy should receive 36 additional weeks of peginterferon alfa plus ribavirin therapy. 4
Dosage reductions or interruptions in therapy with telaprevir should be avoided. 4 If drug-induced adverse events are a problem, the drug should be discontinued or the dose of peginterferon alfa or ribavirin adjusted.
Discontinuation of telaprevir therapy should be considered if the patient has an inadequate response (HCV RNA levels of 1,000 units/mL or more after 4 or 12 weeks of telaprevir plus peginterferon alfa/ribavirin therapy) or confirmed detectable HCV RNA levels at treatment week 24. 4
Telaprevir is not recommended for patients with moderate or severe hepatic impairment (Child-Pugh class B or C, score of 7 or more) or patients with decompensated liver disease; no adjustments in telaprevir therapy are needed in patients with renal dysfunction. However, it has not been evaluated in patients with CrCl 50 mL/min or less. 4
A small study with 161 patients compared the efficacy of telaprevir administered every 8 hours in combination with peginterferon alfa-2a or alfa-2b plus ribavirin versus every 12 hours with the same drug combinations. The results of this study showed no difference in the achievement of SVR between the every-8-hour and the every-12-hour dosing regimen or the type of peginterferon alfa used.55,56 These findings need to be confirmed with a larger study, but may help to decrease the possible poor adherence rate generally seen with 3 times daily drug regimen versus twice-daily drug regimens.
Product Availability and Storage
Telaprevir received US FDA approval on May 23, 2011. 17 It is available as 375 mg tablets in 28-day packs containing 4 weekly cartons of 7 blister strips (6 tablets per blister strip) or as bottles containing 168 tablets. 4
The product should be stored at 25°C (77°F); excursions are permitted between 15°C and 30°C (59°F and 86°F). Once the multiple-dose bottle is opened, it should be kept tightly closed and used within 28 days. 4
Risk Evaluation and Mitigation Strategy (Rems)
No REMS is required for telaprevir. 17
Conclusion
The addition of a 12-week regimen of telaprevir to a standard regimen of peginterferon alfa and ribavirin has exhibited enhanced SVR in patients with genotype 1 hepatitis C without a substantial increase in toxicity. Additional data are necessary to assess the potential for adverse effects and to identify the optimal protocol. In the absence of substantial toxicity, telaprevir is likely to be widely incorporated into treatment regimens for hepatitis C. Boceprevir and telaprevir are both likely to be widely incorporated into treatment regimens for hepatitis C. Comparative data with telaprevir will be valuable, particularly to help discern any differences in the adverse effect profiles of these 2 drugs. At this time, no head-to-head studies have been conducted, and the individual studies with boceprevir and telaprevir have used the agents in conjunction with different pegylated interferons and different ribavirin doses and for differing durations of therapy, which makes direct comparisons difficult.