
Abstract
The complexity of cancer chemotherapy requires pharmacists be familiar with the complicated regimens and highly toxic agents used. This column reviews various issues related to preparation, dispensing, and administration of antineoplastic therapy, and the agents, both commercially available and investigational, used to treat malignant diseases.
Drug Name: Brentuximab vedotin
Synonyms: cAC10-vcMMAE, SGN-35, Adcetris
Mechanism of Action
Brentuximab is an antibody-drug conjugate (ADC) consisting of a CD30 specific IgG1 antibody, a microtubule-attaching drug monomethyl auristatin E (MMAE), and a protease linker that binds these 2 components together. In vitro data suggest the CD30 portion of the drug binds to the cancer cell's CD30 receptor, and the cancer cell admits the drug complex which is cleaved, allowing the MMAE to bind to tubulin. The MMAE shuts down the microtubule mechanism within the cell, inducing cell cycle arrest and eventually cell death.
Pharmacokinetics
The ADC maximum concentration is reached at or near the end of the infusion. The ADC serum concentration declines multiexponentially, with a terminal serum half-life (t1/2) ranging from 4 to 6 days. Exposure to the complex was 1.2 to 2.7 mg/kg. A maximum MMAE concentration is reached between 1 to 3 days. The MMAE portion decreases with continuous administration, with about a 50% to 80% decrease of the first dose being observed at the next dose. The mean steady-state volume of distribution (Vd) of the ADC is 6-10 L in humans. MMAE is a substrate of P-glycoprotein (P-gp) and is 68% to 82% plasma protein bound.
MMAE that is released in the cell is metabolized via oxidation exclusively by CYP3A4/5; no other CYP450 enzymes have been shown to be involved in vitro. Elimination is limited by the rate of release by the ADC complex. Twenty-four percent of the total MMAE administered was recovered in the urine and feces over 1 week, nearly 75% as unchanged drug in the feces. Neither gender, age, nor ethnicity seems to affect the pharmacokinetics.
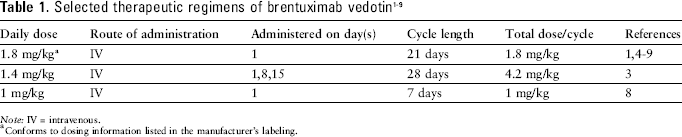
Note: IV = intravenous.
Conforms to dosing information listed in the manufacturer's labeling.
Preparation
Follow institutional policies for preparation of hazardous medications when dispensing brentuximab. Use brentuximab 50 mg vial. Dilute with 10.5 mL sterile water for injection (SWFI) to yield a concentration of 5 mg/mL. Inject the SWFI slowly down the wall of the vial. Swirl the vial; do not shake. Add the drug to at least 100 mL 0.9% sodium chloride (NS), 5% dextrose injection (D5W), or Lactated Ringer's injection to a final concentration between 0.4 mg/mL and 1.8 mg/mL. Gently invert the bag to mix.
Stability
Store the diluted solution at 2° to 8°C [35.6° to 46.4°F], and use within 24 hours. Do not freeze.
Administration
Infuse over 30 minutes.
Toxicities
Drug Name: Vemurafenib
Synonyms: RG7204, PLX4032, R05185426, Zelboraf
Mechanism of Action
Vemurafenib is a small molecule selective inhibitor of the V600E mutation of the BRAF oncogene. Confirm that the patient has the V600E mutation prior to initiating vemurafenib. The US Food and Drug Administration (FDA) mandates use of the Cobas 4800 BRAF V600 mutation test for determining the BRAF mutation. Activated BRAF proteins can cause cell proliferation in the absence of growth factors, leading to abnormal cell proliferation. Vemurafenib interrupts the BRAF/MEK step in the BRAF/MEK/ERK pathway that is exhibited in about 50% of melanoma cells. The inhibition of mutated BRAF serine-threonine kinase, including V600E BRAF, leads to cell death. Nearly all melanoma cells with V600E BRAF respond to vemurafenib.
Pharmacokinetics
Vemurafenib, 960 mg orally (PO) twice daily for 15 days, follows a 1-compartment model with first-order absorption and elimination. The median time to maximum concentration (tmax) following oral administration is 3 hours.
At 960 mg PO twice daily, the mean maximum concentration (Cmax) is 62 ± 17 mcg/mL. The area under the time-concentration curve (AUC) for 0 through 12 hours was 601 ± 170 mcg·h/mL. Vemurafenib is highly bound (greater than 99%) to human albumin and alpha-1 acid glycoprotein. The volume of distribution (Vd) is 106 L, but varies widely. Following oral administration, approximately 94% of the drug is recovered in the feces with a median elimination half-life (t1/2) of 57 hours (range, 30 to 120 hours).
In vitro studies indicate that vemurafenib is a substrate for, and inhibitor of, the efflux transporter P-glycoprotein and a CYP3A4 substrate.

Note: PO = oral.
Conforms to dosing information listed in the manufacturer's labeling. Given as 960 mg twice daily.
Preparation
Follow institutional policies for preparation of hazardous medications when dispensing vemurafenib. Vemurafenib is available in 240 mg film-coated tablets.
Stability
Store at room temperature (20° to 25°C [68° to 77°F]). The tablets are stable until the expiration date on the container.
Administration
Vemurafenib may be taken with or without food. Vemurafenib should be taken in the morning and in the evening, about 12 hours apart. Tablets should be taken whole, with a glass of water.
Toxicities