
Abstract
OBJECTIVE
To review the literature describing the pharmacology, pharmacokinetic properties, efficacy, and adverse effects of prasugrel, a new thienopyridine.
DATA SOURCES
A literature search was conducted (1966–November 2008) of the MEDLINE, Current Contents, EMBASE, and International Pharmaceutical Abstract databases using the key words prasugrel, CS-747, LY640315, and P2Y12. Bibliographies of identified literature were also reviewed for additional references.
STUDY SELECTION AND DATA EXTRACTION
All reports published in English that evaluated prasugrel (or its chemical synonyms) were reviewed. Abstracts without subsequently published reports were excluded.
DATA SYNTHESIS
Given the high rate of recurrent coronary events despite current antiplatelet therapies, agents with potentially greater efficacy are under investigation. Prasugrel is a novel thienopyridine prodrug that is rapidly metabolized to its active platelet-inhibitory metabolite (R-138727) and exerts antiplatelet activity through antagonism of P2Y12 receptors. Prasugrel is very similar in structure and mechanism of action to clopidogrel, as they both possess a methoxycarbonyl group that provides increased pharmacologic activity and an improved hematologic safety profile when compared with ticlopidine. In addition, when compared with clopidogrel, prasugrel demonstrates greater potency and less interpatient variability in the inhibition of platelet aggregation, less in vitro hyporesponsiveness, and, in patients with acute coronary syndromes, a reduced rate of ischemic events. However, this reduction in ischemic events was accompanied by an increased risk of major and fatal bleeding.
CONCLUSIONS
Prasugrel appears to be a promising antiplatelet agent, with emerging clinical data in direct comparison with clopidogrel supporting its role in reducing recurrent ischemic events. Further studies are needed to evaluate the safety and efficacy of prasugrel across various patient populations and clinical scenarios.
THIS ARTICLE IS APPROVED FOR CONTINUING EDUCATION CREDIT ACPE UNIVERSAL PROGRAM NUMBER
407-000-09-001-H01-P
According to the World Health Organization, about one-third of deaths globally each year are secondary to cardiovascular diseases, mainly heart disease and stroke. 1 Thrombus formation on preexisting atherosclerosis is the common feature in these events, with platelet activation and aggregation playing a crucial role in the generation and proliferation of thrombus. As such, aspirin and the thienopyridines have been established as key agents in the acute and long-term prevention of cardiovascular and cerebrovascular events. However, treatment is coupled with an increased risk of bleeding which, in patients at intermediate-to-high risk of vascular events, is generally outweighed by the benefits. 2 Despite the successes of current oral antiplatelet therapies, vascular event rates in clinical trials remain high. 2 One theory exploring this is aspirin and/or thienopyridine resistance.
Aspirin irreversibly inhibits cyclooxygenase-1 (COX-1) in platelets and endothelial cells, preventing the formation of thromboxane A2, which is a potent vasoconstrictor and platelet agonist. Treatment failures may be related to insufficient inhibition of platelet function or true resistance reflected by the failure of aspirin to achieve its pharmacologic effect. 3 Insufficient inhibition of platelet function (while pts. are adherent to therapy) may not be related to actual resistance, but rather to upregulation or increased sensitivity to other mechanisms of platelet activation or thrombus formation. These have been summarized in a review by Cattaneo 3 and include increased sensitivity to adenosine diphosphate (ADP)–induced glycoprotein (GP) IIb/IIIa activation, glycoprotein IIb/IIIa polymorphisms, increased responsiveness to collagen, high plasma levels of von Willebrand factor, synthesis of arachidonic acid derivatives via alternative pathways, and patient factors such as smoking, dyslipidemia, and stress. True aspirin resistance may also exist; proposed mechanisms include decreased bioavailability of aspirin, competition for binding sites (ie, ibuprofen), accelerated platelet turnover, transcellular formation of thromboxane A2 by platelets via prostaglandin histamine2 (H2) released from other cells, thromboxane A2 formation by COX-2, and possible genetic variants of COX-1 that may be less responsive to inhibition by aspirin. Regardless of whether a patient exhibits true aspirin resistance or insufficient inhibition of platelet function secondary to other mechanisms, the presence of a suboptimal response has been associated with an increase in cardiovascular events.4–6
The thienopyridines ticlopidine and clopidogrel are pro-drugs that require hepatic metabolism via CYP3A4 to form their active metabolites. Both drugs irreversibly inactivate the platelet ADP receptor P2Y12, which is required for full platelet activation and aggregation in response to ADP. Treatment failures also occur with thienopyridines, some of which may be due to resistance. Resistance to thienopyridines has been studied mostly with clopidogrel, and potential mechanisms include extrinsic mechanisms such as nonadherence, underdosing, and possibly, drug–drug interactions involving CYP3A4 and the formation of clopidogrel's active metabolite. Intrinsic mechanisms that have been proposed include polymorphisms of the P2Y12 receptor or CYP3A4, increased release of ADP, and increased activity of alternative pathways of platelet activation. 7 The clinical significance of clopidogrel resistance remains to be established, but several studies suggest that it may be associated with an increased risk for recurrent cardiovascular events.8–13
In addition to concerns of resistance, ticlopidine carries a 1% risk of neutropenia and, more rarely (1 case per 1600–5000 pts.), thrombotic thrombocytopenic purpura, both of which may be life threatening. While clopidogrel is not associated with neutropenia, there have been case reports of thrombotic thrombocytopenic purpura, albeit very rare, which also tend to occur early in therapy and require aggressive treatment with plasma exchange. 14
Given the above limitations and concerns with aspirin and the available thienopyridines, the pursuit for antiplatelet agents with a rapid onset of action, predictable platelet suppression, enhanced efficacy, and a favorable safety profile continues. One drug with several of these features is prasugrel, which is the focus of this review.
Data Sources
A literature search was conducted (1966–November 2008) using MEDLINE including in-process and other non-indexed citations, as well as Current Contents, EMBASE Drugs & Pharmacology, and the International Pharmaceutical Abstract databases. Key words included prasugrel, CS-747, LY640315, and P2Y12 receptor antagonist. Bibliographies of identified articles were used to identify additional citations.
Clinical reviews and trials published in English evaluating the pharmacology, pharmacokinetics, and clinical safety and efficacy of prasugrel were selected. Data published in reviews on the role of platelet P2 receptors in hemostasis and thrombosis and their responses to pharmaceutical agents were also included.
Pharmacology
Prasugrel is a novel thienopyridine prodrug that is rapidly metabolized to its active platelet-inhibitory metabolite R-138727 in a 1:1 ratio. Prasugrel is very similar in structure and mechanism of action to clopidogrel, as they both possess a methoxycarbonyl group that provides increased pharmacologic activity and an improved hematologic safety profile compared with ticlopidine (Figure 1).15,16 However, prasugrel appears to be more potent and to have a faster onset of action and longer duration of action when compared with clopidogrel, which may make it more effective in prevention of ischemic events. On the other hand, these features may also increase and/or amplify the bleeding associated with antiplatelet therapy. Prasugrel exerts its antiplatelet activity through its P2Y12 receptor antagonistic properties.17–19
As previously discussed, ADP activates platelets through different ADP receptors, labeled P2 receptors, which are found on the surface of the platelet. The distinct P2 receptors that have been discovered thus far include 2 G protein–complex ADP receptors (P2Y1 and P2Y12) and 1 ligand gated cation channel (P2X), which is activated by adenosine triphosphate. 17
P2Y1 and P2Y12 stimulate platelet aggregation and cause platelet conformational changes. These 2 receptors can also potentiate the actions of other agonists, such as thrombin, collagen, and immune complexes. P2Y1 and P2Y12 play different roles in facilitating hemostasis and thrombosis and have become targets for potential antithrombotic drugs. 17 P2Y12 appears to have a more important role in thrombus formation and stabilization. The role of P2Y12 in the pathogenesis of arterial thrombosis has been described in numerous reports, and it has been demonstrated that P2Y12 antagonists are effective antithrombotic drugs that reduce the risk of adverse cardiovascular events. It is postulated that prasugrel forms a disulfide bridge between the reactive thiol group on R-138727 and the cysteine residue on the P2Y12 ADP receptors and that, by this mechanism, it exerts its long, irreversible P2Y12 blockade activity. 18
Although preclinical data have shown that prasugrel is inactive in platelet aggregation assays in vitro, when given to experimental Sprague-Dawley rats orally, it demonstrated dose-related, potent inhibition of ADP-induced platelet aggregation that was approximately 10 times more potent than that of clopidogrel and 100 times more potent than that of ticlopidine. 18 This enhanced effectiveness is most likely due to prasugrel's ability to powerfully and consistently inhibit ADP-dependent platelet activation.
Pharmacokinetics
With oral administration, prasugrel is completely absorbed and metabolized. 20 This has been described in an open-label human study where a single oral 15-mg dose was given to healthy male subjects. Neither prasugrel nor its hydrolyzed product, R-95913, was detected in the urine or feces of these subjects, indicating full absorption and metabolism. 20 In vivo, prasugrel is rapidly hydrolyzed by esterases to an inactive thiolactone (R-95913), which is then metabolized via one-step oxidation by cytochrome P450, leading to ring opening and the formation of the active metabolite, R-138727. 21 This one-step activation differentiates prasugrel from clopidogrel, which requires a 2-step oxidative process. The one-step process is believed to be responsible for prasugrel's more rapid onset of action, which may be particularly advantageous in the setting of acute coronary syndromes.
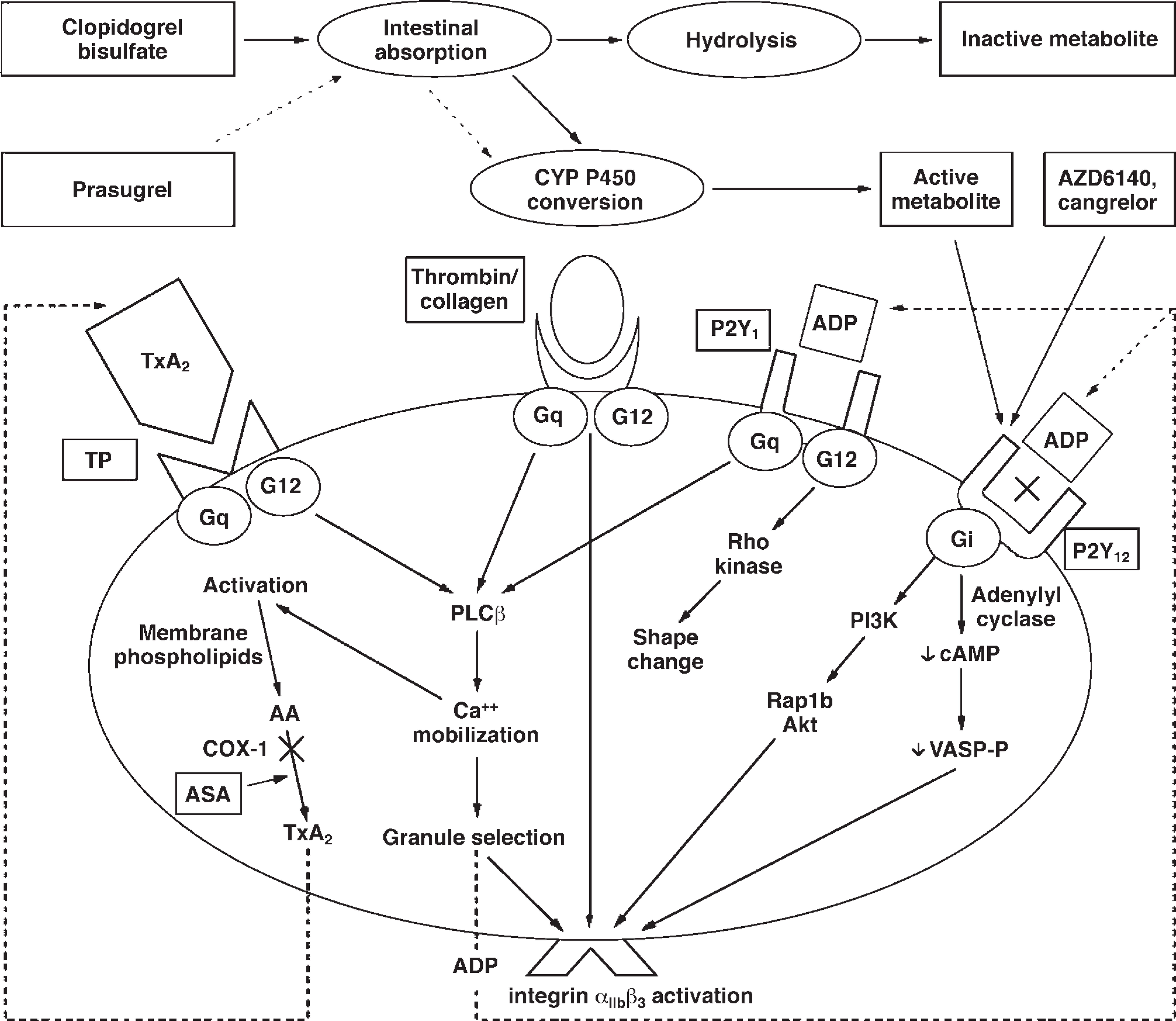
Physiological action of clopidogrel and prasugrel. 15 AA = arachidonic acid; ADP = adenosine diphosphate; ASA = aspirin; cAMP = cyclic adenosine monophosphate; COX = cyclooxygenase; CYP = cytochrome; PLCβ = phospholipase Cβ; P13K = phosphatidylinositol 3-kinase; TP = thromboxane receptor; TxA2 = thromboxane A2; VASP-P = phosphorylated vasodilator-stimulated phosphoprotein. Reprinted by permission from Macmillan Publishers Ltd.: Nature Clinical Practice, 15 copyright 2006.
Prasugrel is converted mainly by CYP3A4; however, unlike clopidogrel, prasugrel is also converted by other cytochrome isoenzymes including CYP2B6, CYP2C9, and CYP2C19. Therefore, it is predicted that, if a single cytochrome P450 isoenzyme involved in R-138727 formation is absent or inhibited, other isoenzymes remain capable of forming the active metabolite. 22 For that reason, it is hypothesized that prasugrel may lead to less interpatient variability and hyporesponsiveness, which may decrease the risk of adverse coronary stent thrombosis and myocardial infarction that is seen despite current antiplatelet regimens. However, data exploring this hypothesis are limited to a randomized trial of 18 healthy subjects who received a 60-mg loading dose (LD) of prasugrel followed by a maintenance dose (MD) of 15 mg daily in conjunction with the potent CYP3A4 inhibitor ketoconazole, at 400 mg/day. Ketoconazole reduced the maximum concentration of R-138727 by 34–48% but did not significantly alter the R-138727 0–24 hour area under the curve after the LD or MD. Prasugrel's ability to inhibit platelet aggregation (≥71%) was essentially unchanged when ketoconazole was coadministered, suggesting that the active compound can indeed be sufficiently formed by other isoenzymes if CYP3A4 is inhibited. 23 Additional studies are needed to confirm this finding.
Prasugrel's onset and duration of action were measured by ex vivo inhibition of ADP-induced platelet aggregation in rat platelet-rich plasma. 23 Prasugrel was found to have a rapid onset of action (<30 min) and a long duration of activity (>3 days). The rapid onset of action may be reflective of the drug's rapid absorption or metabolism to its active compound. This rapid onset of action suggests that prolonged pretreatment with prasugrel may not be necessary; if confirmed, this would favor prasugrel over other available thienopyridines, particularly in the setting of acute coronary syndrome with early percutaneous coronary intervention (PCI). However, the potent and long duration of action may increase the risk of bleeding. The activity that prasugrel exerts 30 minutes after administration is comparable to clopidogrel's peak activity at 6 hours, with a maximal effect of prasugrel seen within 4 hours of dosing.18,24 Despite prasugrel's active metabolite having a half-life of only 3.7 hours, its inhibitory effects do not completely disappear until 96 hours after a dose. This long inhibition of platelet aggregation is equivalent to the life span of the circulating platelets and may reflect the irreversible P2Y12 blockade via the disulfide bridge.18,19
The effects of prasugrel on the liver were measured in a randomized Phase 1 study in which volunteers received prasugrel LD 40 mg/MD 7.5 mg, prasugrel LD 60 mg/MD 15 mg, or placebo for 21 days. 25 Compared with placebo, prasugrel produced no clinically or statistically significant increases in alanine aminotransferase or aspartate aminotransferase, inferring that it will not be hepatotoxic. Because prasugrel is metabolized to an active metabolite by the liver, it is possible that a dosage adjustment may be necessary in patients with hepatic disease, although that has not been established in the current available data. Also, how age, sex, and renal function will affect the pharmacokinetic parameters of this drug has yet to be determined. In the largest trial to date, TRITON-TIMI 38 (Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel—Thrombolysis in Myocardial Infarction), hazard ratios and rates of the primary endpoint on different subgroups, including age, creatinine clearance, and sex, were calculated. 26 Prasugrel displayed significant net benefits versus clopidogrel in patients less than 65 years old, those with a creatinine clearance of 60 mL/min or more, and men. Conversely, patients aged 75 years or more, and those weighing less than 60 kg did not receive overall net benefit from prasugrel, possibly due to a smaller body size or altered disposition of the drug, which may have led to increased production of the active metabolite and increased bleeding. In fact, 2 small studies were recently suspended until protocol amendments can be made to adjust for preliminary results from pharmacokinetic analyses which indicate that a dosage adjustment may be appropriate for certain populations. 27
Phase 1 Trials
There have been multiple Phase 1 trials conducted with prasugrel that have demonstrated rapid, high, and sustained inhibition of ADP-mediated platelet aggregation and acceptable tolerability.25,28,29 Table 1 includes a summary of pertinent Phase 1 trials that directly compared prasugrel with clopidogrel.24,30–32
Clinical Trials
There are currently 4 published Phase 2 trials that compared prasugrel with clopidogrel in patients undergoing PCI or in those with coronary artery disease. The JUMBO-TIMI 26 (Joint Utilization of Medications to Block Platelets Optimally—Thrombosis in Myocardial Infarction) trial investigated the safety and tolerability of prasugrel versus clopidogrel after PCI. 33 This trial was a multicenter, randomized, parallel-group, double-blind, double-dummy comparator-controlled study including patients undergoing elective or urgent PCI with intended coronary stenting. Patients were excluded for several reasons: ST-elevation myocardial infarction (STEMI), high risk of bleeding, and use of proton-pump inhibitor (PPI) (within 12 h or scheduled postprocedure) or H2-blocker therapy (within 2 h prior to PCI). Although possible drug–drug interactions between prasugrel and H2-blocker therapy or PPI may have been considered in this trial, further studies have not revealed interactions between prasugrel and PPIs or H2 blockers. 34 Subsequently, more recent clinical trials have not used this as exclusion criteria.
Subjects were randomized into 1 of 4 groups of oral medication: prasugrel low dose (40 mg LD followed by 7.5 mg daily); prasugrel intermediate dose (60 mg LD and 10 mg daily); prasugrel high dose (60 mg LD and 15 mg daily); or clopidogrel standard dose (300 mg LD and 75 mg daily). The initial dose was given anytime between the times of diagnostic angiogram to when the patient left the recovery room after PCI (average time not reported). Maintenance therapy was continued for 29–34 days. The primary safety endpoint was non–coronary artery bypass graft (CABG)–related “significant hemorrhage” at 30 days, using TIMI standard definitions for combined major and minor hemorrhage. In TIMI standards, major hemorrhage is defined as clinically overt hemorrhage with a hemoglobin drop greater than 5 g/dL. Minor hemorrhage is a clinically overt bleeding episode with a hemoglobin decrease less than 3 g/dL. 35 The trial was not powered to detect differences in efficacy endpoints; rather, composite major adverse cardiac events at a 30-day visit after PCI, along with other efficacy endpoints, were reported (Table 2). 33
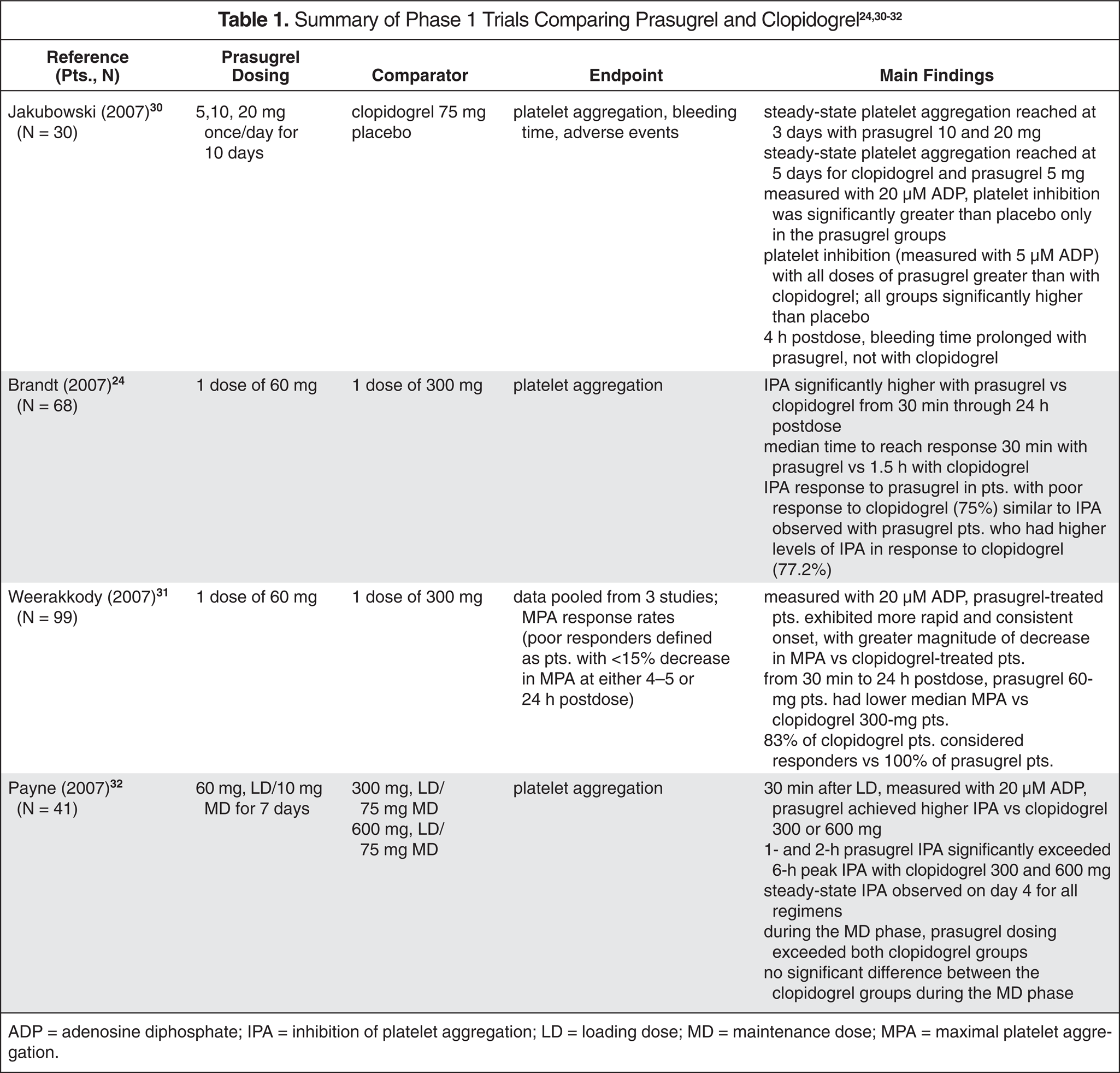
ADP = adenosine diphosphate; IPA = inhibition of platelet aggregation; LD = loading dose; MD = maintenance dose; MPA = maximal platelet aggregation.
This intent-to-treat trial demonstrated no statistically significant differences between prasugrel and clopidogrel in the primary safety endpoint or major adverse cardiac events (Table 2). Bleeding rates were not significantly different; however, the study lacked the power to detect a statistically significant difference in the primary safety endpoint of combined bleeding, as both the prasugrel and clopidogrel arms had lower-than-expected bleeding rates. The study could not exclude a modest increase in bleeding associated with prasugrel. Although the incidence was not statistically significant, 3 deaths occurred in the high-dose prasugrel arm (n = 251); there were no deaths in the clopidogrel arm (n = 254). In addition, 2 nonhemorrhagic strokes in the prasugrel intermediate-dose arm (n = 200) and 1 hemorrhagic stroke within the high-dose arm occurred, whereas none occurred in the clopidogrel arm. However, clinical target vessel thrombosis was significantly higher in the clopidogrel arm (p = 0.024; HR = 0.26; 95% CI–0.07 to 0.92). 33 A subset of this study evaluated 9 patients representing the 4 intervention groups and compared the antiplatelet properties of prasugrel versus those of clopidogrel. 36 When comparing the doses of prasugrel (40 mg/7.5 mg, 60 mg/10 mg, 60 mg/15 mg LD/MD) and clopidogrel (300 mg LD/75 mg MD), the investigators concluded that prasugrel is a more potent antiplatelet agent than is clopidogrel.
Patients with stable coronary artery disease were studied by Jernberg et al. 37 in 2 centers (Sweden and the US). This was a Phase 2, randomized, partially blind, parallel-group study comparing clopidogrel 300 mg LD followed by 75 mg MD with 1 of 4 prasugrel groups: 40 mg LD/5 mg MD, 40 mg LD/7.5 mg MD, 60 mg LD/10 mg MD, or 60 mg LD/15 mg MD for 26–32 days. Subjects also completed a 7- day run-in period with aspirin 325 mg daily, which was continued throughout the treatment period. The trial was double-blind in relation to the prasugrel dose; however, aspirin and clopidogrel were dosed in an open-label manner. Patients were defined as having stable coronary artery disease if they had chronic stable angina, prior history of unstable angina or myocardial infarction, previous coronary revascularization, or disease of at least one coronary vessel demonstrated on previous angiography or noninvasive imaging procedure. Patients were considered unstable if they had unstable coronary artery disease within the previous 30 days or coronary artery intervention within 90 days or planned within the next 40 days. The primary endpoint was inhibition of platelet aggregation (IPA) on day 28. The study also evaluated the rate of nonresponders, with nonresponse defined as the inability to achieve IPA of greater than or equal to 20% during MD administration.
Ninety-nine subjects completed the study. At the 2-, 4-, and 6-hour post-LD evaluation, the IPA was significantly higher for both the 40- and 60-mg LD prasugrel groups when compared with the clopidogrel 300-mg group. The prasugrel MD doses of 10 and 15 mg daily resulted in a significantly higher IPA compared with clopidogrel for days 7 and 28. The lower maintenance doses for prasugrel did not achieve statistical significance compared with clopidogrel. When comparing the LD groups, clopidogrel had a significantly higher rate of nonresponders to 20 μM ADP (52%) compared with both prasugrel LD groups, which had the same rate (3%; p ≤ 0.00002). The 10- and 15-mg MD of prasugrel had 0% nonresponders versus clopidogrel at 45% (p = 0.0007), prasugrel 5 mg at 36%, and prasugrel 7.5 mg at 21%. 37 Some of these results have been questioned, given the discrepancy in Swedish rates to the US rates of response. 38 There was a higher IPA exhibited at one site versus the other; however, the investigators stated that the relative treatment effects observed were the same at each site. This study concluded that prasugrel (40–60 mg LD and 10–15 mg MD) achieved a higher IPA and lower rate of nonresponse when compared with clopidogrel 300 mg LD/75 mg MD. 37
JUMBO-TIMI 26 Primary and Secondary Endpoints 33
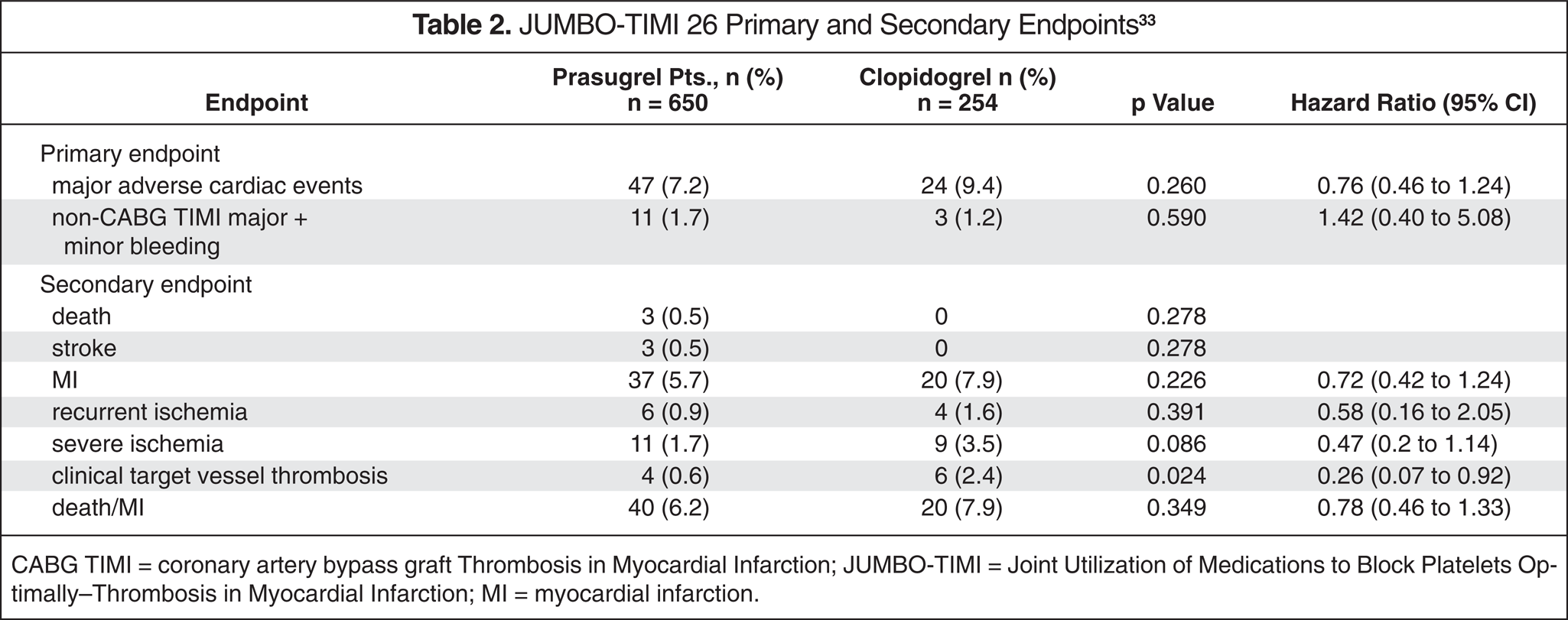
CABG TIMI = coronary artery bypass graft Thrombosis in Myocardial Infarction; JUMBO-TIMI = Joint Utilization of Medications to Block Platelets Optimally–Thrombosis in Myocardial Infarction; MI = myocardial infarction.
Wallentin et al. 39 published a similar Phase 2, randomized, double-blind, double-dummy, 2-arm parallel-group study in patients with stable coronary artery disease. The primary objective of this study was to compare pharmacodynamic and pharmacokinetic effects of prasugrel 60 mg LD and 10 mg daily MD with the higher clopidogrel 600 mg LD and 75 mg daily MD for 28 days. All subjects were given aspirin 75 mg daily for a run-in period of 21 days and throughout the study. The primary outcome of the study was mean change in maximal platelet aggregation to 20 μM ADP at 2 hours post-LD. The study was conducted in 110 patients and reached statistical significance for its primary endpoint, with a change of maximal platelet aggregation of −41.6 for prasugrel and −17.7 for clopidogrel (p < 0.001). A difference was present in the prasugrel group at 30 minutes post-LD and continued throughout the study. This magnitude of effect was not seen with clopidogrel until 2–4 hours post-LD. This trial concluded that, when compared with clopidogrel 600 mg LD and 75 mg daily MD, prasugrel 60 mg LD and 10 mg daily MD provided a faster and greater inhibition of ADP-induced platelet aggregation in patients with stable coronary artery disease.
PRINCIPLE-TIMI 44 (Prasugrel in Comparison to Clopidogrel for Inhibition of Platelet Activation and Aggregation—Thrombolysis in Myocardial Infarction 44), authored by Wiviott et al., 40 was a Phase 2 randomized, double-blind, double-dummy, crossover trial. Subjects were given either high-dose clopidogrel at 600 mg LD/150 mg MD or prasugrel 60 mg LD/10 mg MD for around 15 days. At that time, patients were crossed over into the alternative maintenance therapy for about 14 days. The trial was designed to enroll patients with PCI who were not receiving GP IIb/IIIa inhibitors; however, the LD was given prior to coronary angiography. Because of this, some patients received the LD and did not have PCI and some received a GP IIb/IIIa inhibitor and were excluded from subsequent analysis. The loading dose was to be administered approximately 1 hour prior to when cardiac catheterization (no less than 30 min) was expected to begin. The primary efficacy outcome was IPA at 6 hours and 14 days (pre- and postcrossover). The primary safety measure was non–CABG-related TIMI major or minor bleeding through day 15. There were 197 subjects included in the analyses of this study.
Six hours after LD, there was a significantly greater mean IPA ± SD in the prasugrel group (74.8 ± 13.0%) versus the clopidogrel group (31.8 ± 21.1%; p < 0.0001). In addition, the investigators found a more consistent level of inhibition with the prasugrel group. A favorable decrease in IPA was also found at day 14 for MD therapy (eg, day 15 pre-crossover, day 29 post-crossover). For the primary safety endpoint, no statistically significant difference was found between the 2 groups. The pre-crossover prasugrel group experienced a major or minor bleed in 18.5% (n = 19) of patients versus 14.1% (n = 14) of clopidogrel patients. After the crossover, no patients switching from prasugrel to clopidogrel experienced a hemorrhagic adverse event versus 4 patients who did experience a bleed after switching from clopidogrel to prasugrel. This study concluded that in subjects with planned PCI, prasugrel 60 mg LD/10 mg MD had a more rapid onset and higher, more consistent level of antiplatelet effect compared with subjects who received clopidogrel 600 mg LD/150 mg MD. 40 Currently, one prasugrel Phase 3 investigation, TRITON-TIMI 38, has been published to assess both the efficacy and safety of prasugrel in comparison with clopidogrel. 26
TRITON-TIMI 38 was a double-blind trial designed to evaluate the use of prasugrel versus clopidogrel in patients with moderate- to high-risk acute coronary syndromes and planned PCI. Treatment groups consisted of prasugrel 60 mg LD followed by 10 mg daily MD or clopidogrel 300 mg LD followed by 75 mg daily MD for the duration of the study (up to 15 mo). The loading dose was to be given anytime between randomization and 1 hour after the patient was transferred from the cardiac catheterization laboratory. Before randomization, the patients’ coronary anatomy had to be known before they were considered to be suitable for PCI. If the coronary anatomy was known or primary PCI for STEMI was planned, randomization occurred prior to PCI and the study drug was given as soon as possible after randomization. For subjects with unstable angina or non-STEMI (NSTEMI) or for those who enrolled after medical treatment for STEMI, coronary anatomy was verified for patients to be considered suitable for PCI prior to randomization and study drug administration. During this intervention, patients were also required to be taking 75–162 mg of aspirin daily. The primary efficacy endpoint was a composite of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke. Key safety endpoints included TIMI major bleeding, TIMI life-threatening bleeding, and TIMI major and minor bleeding.
The study randomized 13,608 patients with either unstable angina (UA)/NSTEMI or STEMI and evaluated patients for a median duration of 14.5 months. Study drug was received by 25% of patients prior to placement of the first coronary guidewire (26% prasugrel, 25% clopidogrel); by 74% of patients after first guidewire or within 1 hour of PCI (73% prasugrel, 74% clopidogrel); and by 1% of patients (1% in both groups) more than 1 hour post-PCI. The average time of study drug LD in relationship to PCI was not provided. The primary efficacy endpoint was significantly reduced in the prasugrel group for the UA/NSTEMI (hazard ratio [HR] 0.82; 95% CI 0.73 to 0.93; p = 0.002) and the STEMI (HR 0.79; 95% CI 0.65 to 0.97; p = 0.02) groups. A significant reduction in the primary endpoint was seen at the first prespecified point of 3 days and persisted throughout the study period. Within this composite endpoint, the difference was primarily attributed to the reduction in fatal and nonfatal myocardial infarction in the prasugrel arm. There was no significant difference between the 2 groups in the rate of stroke or death from cardiovascular causes (other than myocardial infarction). Among prespecified subgroups, prasugrel demonstrated benefit in patients with (HR 0.79; 95% CI 0.69 to 0.91; p < 0.001) or without (HR 0.84; 95% CI 0.72 to 0.99; p = 0.03) use of glycoprotein IIb/IIIa inhibitors. The following patient groups also achieved statistically significant benefits when viewed separately: males, patients less than 65 years old, patients with or without diabetes, those receiving a bare-metal or drug-eluting stent, and patients with a creatinine clearance greater than or equal to 60 mL/min. Patient subgroups that did not achieve a statistically significant difference between prasugrel and clopidogrel included females, as well as all patients (male or female) 65 years of age or more. However, these are all subgroup analyses and subject to the inherent limitations with such analyses. 26
Differences found in the safety endpoints favored clopidogrel. There was a significant increase in the number of patients with at least one episode of TIMI major bleeding in the prasugrel group versus the clopidogrel group (2.4% vs 1.8%, respectively; HR 1.32; 95% CI 1.03 to 1.68; p = 0.03), including a higher rate of life-threatening bleeding in the prasugrel group (HR 1.52; 95% CI 1.05 to 2.44; p = 0.03). Fatal TIMI major bleeding occurred more in the prasugrel group (0.4%) compared with the clopidogrel group (0.1%). Intracranial hemorrhage rates were the same for both groups (0.3%), and more patients discontinued the study drug because of hemorrhage (prasugrel 2.5% vs clopidogrel 1.4%; p < 0.001). When efficacy and bleeding endpoints were combined, net clinical benefits (death from any cause, nonfatal myocardial infarction, nonfatal stroke, and TIMI major hemorrhage) favored prasugrel in the overall study population, but not in all patient subgroups (HR 0.87; 95% CI 0.79 to 0.95; p = 0.004). Due to these disparities in efficacy versus safety endpoints, further post hoc analysis was conducted that defined net clinical benefit as rate of death from any cause, nonfatal myocardial infarction, nonfatal stroke, or non–CABG-related nonfatal TIMI major bleeding associated with the use of prasugrel, or patients who had net harm. Identified groups not experiencing a benefit or possibly experiencing harm from prasugrel included patients with previous stroke or transient ischemic attack, those weighing less than 60 kg, and patients greater than 75 years old. These patients were also identified as having higher rates of bleeding. In patients without any of these 3 risk factors, there was greater efficacy with prasugrel and no significant difference in rate of major bleeding. 26
This trial provides some evidence favoring the use of prasugrel over clopidogrel in patients with acute coronary syndromes. However, this comes at a cost of increased serious bleeding and loss of net clinical benefit, which may be heightened in certain subgroups, as described previously. Limitations of this study include concerns over the amount and timing of the LD. The LD of clopidogrel in relation to PCI was not provided; however, 75% of patients received the LD after the first guidewire was placed. In contrast, the CREDO (Clopidogrel for the Reduction of Events During Observation) trial studied the administration of a 300-mg LD of clopidogrel 3–24 hours prior to PCI, with or without GP IIB/IIIa inhibitors. A subgroup analysis of this study suggested that a 300-mg LD of clopidogrel was more efficacious when given further (at least 6 h) in advance of PCI. 41 The ARMYDA-2 (Antiplatelet Therapy for Reduction of Myocardial Damage During Angioplasty) trial studied clopidogrel 300 mg versus 600 mg taken 4–8 hours prior to PCI and concluded that a clopidogrel LD of 600 mg was more beneficial, with similar safety endpoints, than clopidogrel 300 mg when administered 4–8 hours prior to PCI. 42 The European Society of Cardiology guidelines recommend a clopidogrel LD of 600 mg in patients who have not received an LD of clopidogrel the day before planned PCI. 43 Interestingly, in the TRITON-TIMI 38 trial, the study event rate was driven by periprocedural myocardial infarction, which may partly be explained by inadequate exposure to clopidogrel prior to PCI. Furthermore, cumulative Kaplan-Meier estimates of the primary endpoint during the follow-up period diverged early (within 3 days), with little, if any, divergence afterwards. This may also be suggestive that the majority of positive effect was early, when exposure to clopidogrel prior to PCI may have been suboptimal. In contrast, bleeding (in contrast to benefit) was not realized early in the trial and continued to increase throughout the trial duration.
Wiviott et al. 44 published a substudy examining the rates of ischemic events and stent thrombosis in TRITON-TIMI 38 and the size and timing of the effects of prasugrel compared with those of clopidogrel in patients receiving different types of intracoronary stents. Although this was a substudy, 94% of patients from the TRITON-TIMI 38 trial received stents and therefore were included. Prasugrel's benefits toward the primary endpoint of cardiovascular death/nonfatal myocardial infarction/nonfatal stroke were observed in patients with at least one stent (prasugrel 9.7% vs clopidogrel 11.9%; HR 0.81 [95% CI 0.72 to 0.90]; p = 0.0001), in patients who received a bare-metal stent only (prasugrel 10% vs clopidogrel 12.2%; 0.80 [0.69 to 0.93]; p = 0.003), and in patients who received a drug-eluting stent only (prasugrel 9% vs clopidogrel 11.1%; 0.82 [0.69 to 0.97]; p = 0.019). In addition, stent thrombosis was significantly reduced in patients who received at least one stent (irrespective of type), in those who received a bare-metal stent only, and in patients who received a drug-eluting stent only.
Two additional subanalyses from the TRITON-TIMI 38 trial have recently been published. Murphy et al. 45 evaluated the effects of prasugrel versus clopidogrel on recurrence of the primary efficacy endpoint (cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke). They observed that prasugrel not only decreased first occurrence of this endpoint, but also decreased recurrence, thus, decreasing total number of events (RR 0.79, 95% CI 0.71 to 0.87; p < 0.001). Wiviott et al. 46 published a substudy evaluating net benefits in patients with diabetes. Of the 13,608 patients in the major trial, 3146 (23%) had a preexisting history of diabetes. Prasugrel reduced the primary endpoint in patients with diabetes (12.2% vs 17.0%, HR 0.70, 95% CI 0.58 to 0.85; p < 0.001) and without diabetes (9.2% vs 10.6%; HR 0.86; p = 0.02); however, the difference between patients with diabetes versus those without diabetes did not reach statistical significance (pinteraction = 0.009). The number of patients with diabetes needed to treat with prasugrel to prevent one primary endpoint was 21 versus 71 patients without diabetes. Similar rates of TIMI major hemorrhage were observed in prasugrel patients with or without diabetes; therefore, a trend toward an increase in net clinical benefit was observed (with diabetes 14.6% vs 19.2%; HR 0.74; p = 0.001; without diabetes 11.5% vs 12.3%; HR 0.92; p = 0.16, pinteraction = 0.05). However, both of these are subanalyses and thus subject to inherent limitations with these types of investigations.
Precautions and Contraindications
The greatest insight into possible precautions and contraindications with prasugrel use can be gathered from the large randomized trials that have evaluated it. Based on the exclusion criteria in these trials, prasugrel may need to be avoided or cautiously used in patients with the following risk factors: fibrinolytic therapy within 24 hours, bleeding risk, stroke within 2 years, intracranial neoplasm, atrioventricular malformation, aneurysm, uncontrolled hypertension, and concomitant anticoagulation therapies.33,47,48 TRITON-TIMI 38 suggested that there also may be a subset of patients who are at an increased risk for serious bleeding after prasugrel use. 26 A post hoc analysis on different subgroups revealed an increased risk for major and fatal hemorrhage among patients with prior stroke or transient ischemic attacks, those aged 75 or older, and those who weigh less than 60 kg. The post hoc analysis also suggested that people with a history of cerebrovascular events were not only at a higher risk for major or fatal bleeds, but also had no net clinical benefit from prasugrel, indicating significant net harm from its use. 40 This is not entirely surprising, as the MATCH (Management of Atherothrombosis with Clopidogrel in High-Risk Patients with Recent Transient Ischemic Attacks or Ischemic Stroke) trial found an increase in intracranial bleeding among patients with a history of stroke or transient ischemic attacks who were receiving aggressive antiplatelet therapy. 49 Further studies need to be conducted to determine whether these 3 subsets of patients will benefit from a dosage reduction or possibly need to avoid the use of prasugrel.
Given that prasugrel is very similar to clopidogrel and ticlopidine, the precautions and contraindications to those drugs should be considered until more data on prasugrel are available. Clopidogrel is contraindicated in patients with active bleeding (eg, peptic ulcer or intracranial hemorrhage). Caution should be used in patients on clopidogrel who have thrombotic thrombocytopenic purpura, risk of bleeding secondary to trauma, severe liver disease, or severe renal impairment. Clopidogrel should also be discontinued 5 days before elective surgery if antiplatelet effects are unwarranted, given that the life span of a platelet is 7–10 days. 50 Prasugrel and clopidogrel both irreversibly inhibit platelet aggregation, with long durations of action, and there may be similar or additional requirements for prasugrel use prior to surgery.
Drug Interactions
Based on prasugrel's pharmacokinetics, one may hypothesize that drugs that affect CYP3A may significantly alter concentrations of the active metabolite, R-138727. However, it has been demonstrated that prasugrel also undergoes oxidation to its active metabolite via other isoenzymes, making therapeutic failure less likely. Small et al. 34 assessed the pharmacokinetic and pharmacodynamic effects of single-dose prasugrel (60 mg) and clopidogrel (300 mg) when given with lansoprazole 30 mg daily. Decreases in area under the curve and maximum concentration were demonstrated in subjects in both the prasugrel and clopidogrel groups; however, a decrease in IPA was not observed in prasugrel patients. At 24 hours postdose, patients who received only clopidogrel reached a higher mean platelet inhibition when compared with the clopidogrel plus lansoprazole group (49% vs 39%, respectively; p = 0.046). Furthermore, it has been concluded that neither R-95913 nor R-138727 substantially inhibit the most common isoenzymes CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. 51 Prasugrel was also found to insignificantly inhibit CYP2B6 in a study that assessed CYP2B6 inhibition by prasugrel using bupropion as a probe substrate. 48 Thus, it has been postulated that prasugrel does not require a dosage adjustment when given concomitantly with drugs that are metabolized via the cytochrome P450 system.22,48
We can hypothesize, based on the pharmacology of prasugrel, that aspirin and other anticoagulants (ie, warfarin, heparin, thrombolytics), as well as concomitant use with other antiplatelets, would increase the risk of bleeding. In an open-label, randomized study, prasugrel at 4 different doses (LD/MD: 20/5 mg, 30/7.5 mg, 40/10 mg, or 60/15 mg) was coadministered with aspirin 325 mg to 45 healthy subjects between 18 and 60 years old. 52 There were no clinically significant changes in the hematologic parameters; thus, one may conclude that aspirin and prasugrel can safely be administered concomitantly. However, as mentioned previously, in TRITON-TIMI 38 there were significantly more major, life-threatening, fatal, and intracranial bleeds in the prasugrel group when compared with clopidogrel (both added to background therapy with aspirin). 26 This finding requires clinicians to carefully weigh a patient's risk for bleeding against potential benefit. Even though more intensive platelet inhibition with prasugrel appears to reduce ischemic events in acute coronary syndromes, one must be cognizant of the increased risk of bleeding seen in certain subgroups of patients, especially those with a history of cerebrovascular events.
Safety and Tolerability
As would be expected, the major adverse event of concern with prasugrel is bleeding, which includes not only major bleeding, but also bleeding that may lead to premature discontinuation of the drug. In TRITON-TIMI 38, more prasugrel patients (2.5%) versus clopidogrel patients (1.4%) discontinued the study drug because of adverse events related to hemorrhage (p < 0.001). 26 As described above, although efficacy in TRITON-TIMI 38 was improved in the prasugrel group compared with the clopidogrel group, bleeding events also exhibited an increase. Increased bleeding was seen in several bleeding categories in prasugrel patients when compared with clopidogrel patients (Table 3). The rate of intracranial hemorrhage was not found to be higher with prasugrel versus clopidogrel; however, post hoc analysis revealed that this rate was increased in subjects with a history of previous stroke or transient ischemic attack. Given this line of risk versus benefit, further post hoc subgroups were evaluated, and patients either with a history of stroke or transient ischemic attack, who were greater than 75 years old, or weighing less that 60 kg, were identified as not realizing the net benefit (or experiencing harm) from prasugrel therapy. Other adverse events reported in the TRITON-TIMI 38 included thrombocytopenia (no significant difference between clopidogrel and prasugrel), neutropenia (higher in clopidogrel), and colonic neoplasms (higher in prasugrel group). With extrapolation based on data from clopidogrel and ticlopidine, a possibility of purpuric disorder, rash, abdominal pain, diarrhea, constipation, dyspepsia, gastritis, arthralgia, back pain, dizziness, headache, agranulocytosis, anemia, neutropenia, thrombotic thrombocytopenic purpura, abnormal renal function, and elevated liver function tests could occur with prasugrel treatment.
Rebound activation of platelets has been a recent concern with clopidogrel, and there has been one published case report of a patient who participated in the JUMBO trial and experienced this adverse event with prasugrel.53,54 A 48-year-old man randomized to receive the 60-mg LD and 10-mg MD of prasugrel experienced significant inhibition of platelet activity, as shown on monitoring at baseline, 4, and 24 hours. However, upon follow-up at 30 days, rebound activity was indicated in all platelet measures, placing the patient above findings in healthy controls. Upon further interview, the patient admitted to discontinuing prasugrel and aspirin shortly after discharge. This case report demonstrates the dangers of nonadherence to antiplatelet therapy and suggests that not only are the benefits decreased in these patients, but that rebound activation may actually place them at even higher risk of negative cardiovascular outcomes.
Dosing and Administration
Data from Phase 1 and 2 studies appear favorable for the 60-mg LD followed by a 10-mg daily MD. The TRITON-TIMI 38 study did report higher efficacy with this dose when compared with the clopidogrel 300-mg LD followed by the 75-mg MD. 26 In addition, the Phase 2 study, PRINCIPLE-TIMI 44, which evaluated antiplatelet activity with prasugrel 60 mg LD/10 mg MD versus higher doses of clopidogrel (600 mg LD/150 mg MD), suggested that this dose may be more efficacious when compared with the higher doses of clopidogrel. 40 Another Phase 3 trial may be helpful in further assessing this. Although this regimen has been used consistently in additional trials, there is some question as to whether a lower prasugrel dosage (or dosage adjustment based on weight or other patient characteristics) would maintain effectiveness while decreasing bleeding.
Bleeding-Related Adverse Events Observed in TRITON-3826
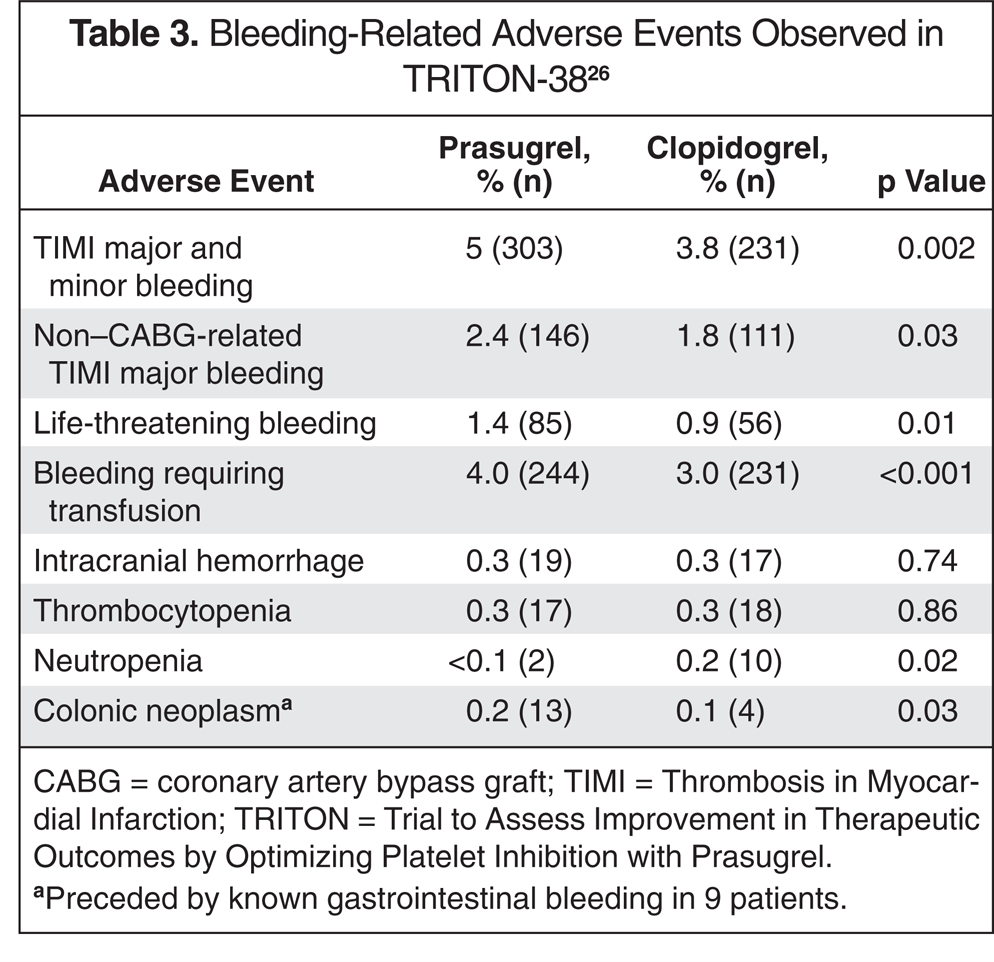
CABG = coronary artery bypass graft; TIMI = Thrombosis in Myocardial Infarction; TRITON = Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel.
Preceded by known gastrointestinal bleeding in 9 patients.
At this time, there are no recommendations regarding dosing adjustment; however, pharmacokinetic and pharmacodynamic studies indicate that it is unlikely that adjustment is needed in patients on medications that affect the cytochrome P450 system or in those with genetic differences in P450 enzyme activity. The TRITON-TIMI 38 study did not mention direct exclusion of patients with decreased renal function; subgroup analysis indicated that patients with a creatinine clearance less than 60 mL/min as well as those greater than 65 years of age were enrolled. 26 Further studies may provide more information regarding exclusion of these patients for treatment candidacy or need for change of dose. In addition, as previously mentioned, 2 small studies were recently suspended until protocol amendments can be made to adjust for preliminary results from pharmacokinetic analyses; this indicates that a dose adjustment may be appropriate for certain populations. 27
The ongoing Phase 3 trial, TRILOGY ACS (Targeted Platelet Inhibition to Clarify the Optimal Strategy to Medically Mange Acute Coronary Syndromes) trial, should provide additional information regarding the subgroups in question. This trial is designed to compare prasugrel with clopidogrel in the medical management of over 10,000 patients with ACS and will be using a reduced dose in certain subgroups based on age and weight. 55 The study is expected to be complete in early 2011. 56
Cost
As of October 1, 2008, there was no information on cost for prasugrel.
Expected Availability
Lilly submitted the New Drug Application for prasugrel in December 2007 and received priority review status by the Food and Drug Administration (FDA) in late February 2008. As of October 1, 2008, the FDA elected to delay its decision on prasugrel and continue its review of the drug. It is currently proposed that prasugrel will be marketed under the name Effient.
Summary
Prasugrel is a novel thienopyridine that demonstrates greater potency and less interpatient variability in the inhibition of platelet aggregation, less in vitro hyporesponsiveness, and, in a large Phase 3 trial in patients with acute coronary syndromes, a reduced rate of ischemic events when compared with clopidogrel. However, this reduction in ischemic events was accompanied by an increased risk of major and fatal bleeding, which necessitates further studies to evaluate the safety and efficacy of prasugrel across various patient populations and clinical scenarios.
The ongoing Phase 3 trial TRILOGY ACS may help address these patient population and clinical scenario concerns. Other dosing strategies should also be explored, including the possibility of using the favorable early pharmacokinetic and pharmacodynamic effects to justify early use of prasugrel followed by longer-term use with a less potent (and subsequently, with less bleeding) agent such as clopidogrel. Only after additional investigations will the role of prasugrel within the antiplatelet armamentarium be fully defined.