
Abstract
Objective
After introducing a new descriptive recording system for congenital craniofacial abnormalities in The Netherlands, common oral clefts are highlighted.
Design
Prospective observational study.
Setting
Fifteen cleft palate teams, united in the Dutch Association for Cleft Palate and Craniofacial Anomalies, registered patients from 1997 to 2006.
Patients
All unoperated patients with a common oral cleft were included.
Main Outcome Measures
Detailed information and birth prevalence rates of cleft lip/alveolus, cleft lip/alveolus and palate, and cleft palate were provided, relating referral age, gender, family history, additional congenital abnormalities, and syndrome diagnoses to these three categories.
Results
This study included 3512 patients, resulting in an overall prevalence of 16.6 per 10,000 live births. Patients showed a cleft lip/alveolus (28%), a cleft lip/alveolus and palate (39%), or a cleft palate (33%). The three categories exhibited very heterogeneous cleft types. Mean referral age was 5.8 months (median 3 weeks). Birth weight was the lowest in cleft palate patients (3238 g; p < .001 to .009). Cleft palate patients showed less positive family history concerning congenital anomalies (23%, p < .001 to .013), but more syndrome diagnoses were established in this category (24%, p < .001). Ten percent of all cleft patients showed additional abnormalities of the head and neck area, and 13% displayed congenital anomalies of other systems.
Conclusions
This new recording method allows adequate description of common oral clefts. Many cleft types exist within these three categories and should be differentiated, because they originate from different time frames and/or cell biological mechanisms during embryogenesis.
Registration and classification of congenital anomalies in general, and common oral clefts in particular, is of paramount importance to provide a solid basis for epidemiologic, clinical, and/or fundamental research. Several registration and classification systems have been developed in order to consistently categorize the observed types of common oral clefts (Kernahan and Stark, 1958; Harkins et al., 1962; Kernahan, 1971; Sandham, 1985; Kriens, 1990; Friedman et al., 1991; Schwartz et al., 1993; Koch et al., 1995; Smith et al., 1998; Ortiz-Posadas et al., 2001; Koul, 2007; Liu et al., 2007). These systems provide details about the cleft types according to anatomic appearance and adequately describe the more frequent variations. However, infrequent types of clefting often cannot be classified, except for the classification of Kriens (1990), adjusted by Koch et al. (1995), which is very time-consuming to complete. Therefore, we have developed a new recording system on behalf of the Dutch Association for Cleft Palate and Craniofacial Anomalies (NVSCA), and the system is embedded in the working group “Registration.” This system, the NVSCA registration, records the individual abnormalities of the primary (lip and alveolus) and secondary palate (hard and soft palate including the uvula) that form the common oral cleft. To anticipate all conceivable abnormalities, the system is based on embryology, morphology, and topography. Furthermore, all other individual craniofacial abnormalities can be described, including, for example, all types of craniosynostosis and congenital ear anomalies. The cleft types are not categorized or coded in any way because this would lead to loss of information. Additional congenital abnormalities of other systems of the body can be indicated as well.
After importing the data in a specific developed computer program, categorization may follow depending on proposed queries; specific categorization may be necessary for specific queries.
Since 1997 all unoperated patients who were referred for assessment of an oral cleft have been recorded nationwide. In The Netherlands, almost every child with an oral cleft is referred to a cleft palate team.
It should be noted that this system is an anonymous recording system. For studies using anonymous data, approval of the NVSCA board is needed; for patient identification, approval of all teams concerned is also required. The patient identification number of the cleft palate team, the birth date, and gender can be used to find the patient's hospital records.
This paper introduces the new recording system and reports the results of 10 years of recording common oral clefts in The Netherlands. Gender, race, referral age, adoption/foster status, birth weight, gestational age, family history, other congenital anomalies, and syndrome diagnoses/sequences/associations were related to the three common categories, cleft lip/alveolus, cleft lip/alveolus and palate, and cleft palate. Detailed information and birth prevalence rates were presented for the three categories of common oral clefts.
Materials and Methods
Patients and NVSCA Recording Form
In 10 years (1997 to 2006), 3512 unoperated patients were referred for the first time to a multidisciplinary consultation for assessment of a cleft lip and/or alveolus and/or palate. After careful examination, these patients were recorded using the NVSCA recording form. The form is well organized (one page only) and is fast and easy to complete. It is composed of three parts: (1) a general section (e.g., ethnic origin), (2) a section for craniofacial abnormalities including common oral clefts, and (3) a section for any congenital abnormality of other parts of the body (Fig. 1). A manual is available (Fig. 2). The section for craniofacial abnormalities includes common oral clefts. In a two-dimensional table, the X axis shows topography (e.g., lip, pre/max, i.e., alveolus and hard palate), and on the Y axis, morphology is depicted (e.g., complete, incomplete, and submucous clefts). In addition, absent (agenesis), malformed (aplasia), and undersized (hypoplasia), or oversized (hyperplasia) parts of the lip, alveolus, and palate can be noted in section 2, as well as all other craniofacial malformations.

Recording form of the NVSCA registration. Reproduced by kind permission of the Department of Plastic and Reconstructive Surgery, Erasmus MC-University Medical Center Rotterdam, The Netherlands.

Manual for the NVSCA registration form.
The authors assessed all forms, and members of the cleft palate teams provided additional information if necessary. In this study, only common oral clefts with or without associated abnormalities were included. Median cleft lip and atypical clefts were excluded for their different pathogenesis.
Gender, race, referral age, adoption/foster status, birth weight, gestational age, family history, other congenital anomalies, and syndrome diagnoses/sequences/associations were related to the three common categories. We assume that the cleft palate teams assess nearly all patients with a common oral cleft, and therefore we were able to produce birth prevalence rates (live births). These birth prevalence rates were computed using general data from the Dutch Central Bureau of Statistics (CBS) and the birth dates of the patients.
Statistical Analysis
Statistical analysis was performed using chi-square test for dichotomous variables (gender, race, adoption/foster status, family history, other congenital anomalies, syndrome diagnoses). Independent samples t test was used for continuous variables (gestational age and birth weight), p values below 5% were considered to be statistically significant. Statistics were performed using a software package (SPSS v 14.0).
Results
General Information
Two thousand fifty male and 1462 female patients were recorded (ratio male/female = 1.40). The common oral clefts were subdivided into three categories: cleft lip/alveolus (CL/A), cleft lip/alveolus and palate (CL/AP), and cleft palate (CP).
The parents were Caucasian in 2943 cases and not Caucasian in 352. The remaining 217 patients had parents with a mixed race (147) or with an unknown race (70). In 75 cases the parents were related to each other (any degree of relationship). Forty-one couples were cousins (second-degree relatives), of which 21 were Caucasian (0.7% of all Caucasian parents), and 20 were non-Caucasian (6% of all non-Caucasian parents). Eighteen couples had a third-degree relationship, and in 16 couples the degree of relationship was unknown.
Mean age at referral was 5.8 months, and the median referral age was 21 days (range 0 days to 43 years). Nine percent of all cleft patients were referred older than 1 year (n = 299). Late referral age was evaluated using the following parameters: adoption/foster status and cleft category. One hundred forty-three patients were adopted/foster children (mean referral age = 18 months). Seventy-eight of the 143 adopted/foster children with a common oral cleft were referred after 1 year of age (55%). This is significantly more than the 221 of the 3327 nonadopted patients (p < .001, mean referral age of all nonadopted patients = 5 months). Significantly more adopted CP patients visited a cleft palate team later than 12 months of age compared with CL/A and CL/AP patients (both p < .001); CL/A and CL/AP adopted patients also differed in referral age (p = .011; Table 1).
Mean birth weight was 3294 g (range 780 g to 5612 g). CL/A and CL/AP patients had a significantly higher birth weight than CP patients (p < .001 and p = .009, respectively; mean CL/A 3364 g, CL/AP 3291 g, and CP 3238 g). However, if patients with additional congenital anomalies of the head and neck area and/or other parts of the body were excluded, no significant differences in birth weight were observed (mean CL/A 3393 g, CL/AP 3331 g, and CP 3326 g; p = .200 to .700).
Referral Age (in Years) of Nonadopted and Adopted Patients Related to Category of Cleft *
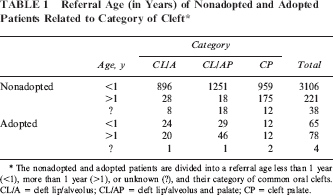
The nonadopted and adopted patients are divided into a referral age less than 1 year (<1), more than 1 year (>1), or unknown (?), and their category of common oral clefts. CL/A = cleft lip/alveolus; CL/AP = cleft lip/alveolus and palate; CP = cleft palate.
Mean gestational age was 39 weeks (range 26 to 43 weeks). No significant differences were observed among patients with CL/A, CL/AP, or CP (p = .190 to .959).
Occurrence of any congenital anomaly among relatives was present in 26% of the cases. More patients with a CL/A or CL/AP (27% and 29%, respectively) showed a positive family history than the CP patients (23%; p = .013 and p < .001, respectively). Twenty-one percent of the patients showed a positive family history for common oral clefts (22% of the CL/A patients, 24% of the CL/AP patients, and 16% of the CP patients). In the CP group, significantly fewer patients with a positive family history for common oral clefts were observed when compared to CL/A or CL/AP (both p < .001), whereas no significant difference was observed between CL/A and CL/AP patients (p = .329).
Types of Common Oral Clefts Within the Three Categories
All frequent cleft types could be easily recorded. For instance, the recording of a right-sided complete cleft lip and alveolus (Fig. 3A); a right-sided complete cleft lip, alveolus, and hard palate, and a complete cleft of the soft palate (Fig. 3B); and an incomplete cleft of the palatum molle and a complete uvular cleft are shown in Figure 3C. Infrequent or unique clefts could also be described such as a left-sided incomplete cleft of the lip with a submucous component, combined with an ipsilateral incomplete alveolar cleft (Fig. 4A); a complete cleft lip on the left side, a complete cleft lip and alveolus on the right side, a left-sided incomplete cleft of the hard palate with a anterior submucous part, a right-sided complete cleft of the hard palate, and a complete cleft of the soft palate (Fig. 4B); or a median submucous cleft of the hard palate and a complete cleft of the soft palate (Fig. 4C).
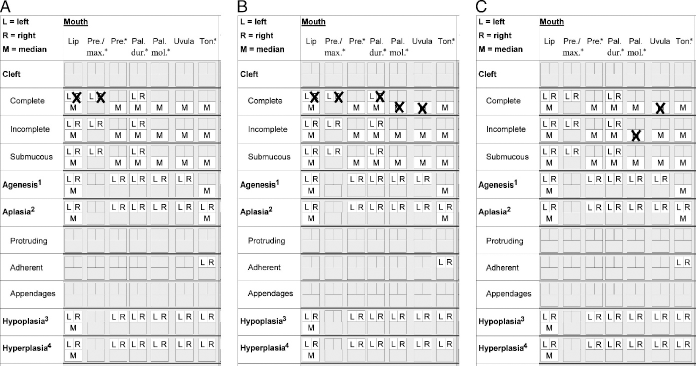
A: A fast and easy recording of a right-sided complete cleft lip and alveolus. B: A right-sided complete cleft lip, alveolus, and hard palate, and a complete cleft of the soft palate. C: An incomplete cleft of the palatum molle and a complete uvular cleft.
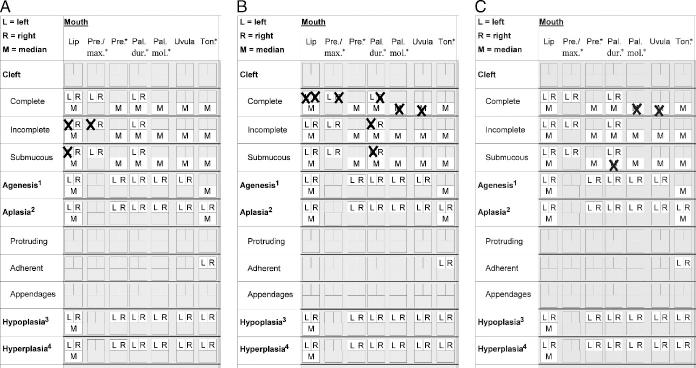
Examples of recording infrequent or unique clefts. A: A left-sided incomplete cleft of the lip with a submucous component, combined with an ipsilateral incomplete alveolar cleft. B: A complete cleft lip on the left side, a complete cleft lip and alveolus on the right side, a left-sided incomplete cleft of the hard palate with an anterior submucous part, a right-sided complete cleft of the hard palate, and a complete cleft of the soft palate. C: A median submucous cleft of the hard palate and a complete cleft of the soft palate.
Nine hundred seventy-seven patients had CL/A (28%). The male/female ratio was 1.72. The most common types of CL/A were incomplete cleft lip (374 patients), incomplete cleft lip and alveolus (267 patients), and complete cleft lip and alveolus (133 patients) (Table 2). Incomplete cleft lip with or without an incomplete alveolar cleft was on the left side in 64%, on the right side in 29%, and bilateral in 7%. A complete cleft lip and alveolus showed left, right, and bilateral involvement in 56%, 31%, and 13%, respectively. Furthermore, 39 and 38 cases exhibited complete cleft lip and incomplete cleft alveolus, and complete cleft lip, respectively, and another 126 patients showed 20 infrequent cleft types (data not shown).
The Most Common Types of CL/A (Gray), CL/AP (Dark Gray), and CP (Light Gray); Left- and/or Right-Sided Clefts Are Not Distinguished Here 1
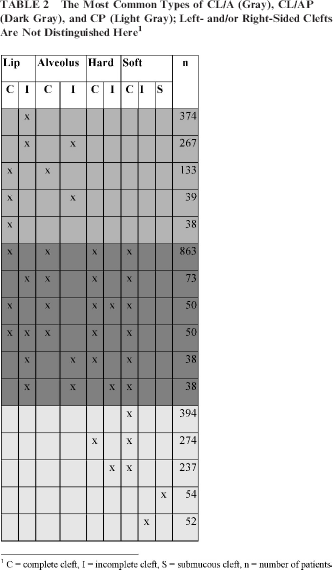
C = complete cleft, I = incomplete cleft, S = submucous cleft, n = number of patients
One thousand three hundred sixty-three patients showed a CL/AP (39%) with a male/female ratio of 2.04. Most frequently, a complete cleft of the lip, alveolus, and palate was observed (863 patients, 63%) (Table 2). Of these cases, 384 clefts of the lip and alveolus were located at the left side (44%), 222 patients showed a cleft lip/alveolus on the right side (26%), and bilateral involvement of the lip and alveolus was present in 257 patients (30%). In addition to the less frequent CL/AP types indicated in Table 2, 251 patients showed 86 infrequent cleft types (data not shown).
One thousand one hundred seventy-two patients showed a CP (33%). The male/female ratio was 0.79. Most frequently, a complete cleft of the soft palate was observed (394 patients, 34%), followed by a complete cleft of the hard and soft palate (274 patients, 23%) and an incomplete cleft of the hard palate combined with a complete cleft of the soft palate (237 patients, 20%). In addition, 54 and 52 patients (Table 2) exhibited a submucous and incomplete cleft of the soft palate, respectively, and another 161 patients showed 30 infrequent types of CP (data not shown).
Additional Abnormalities
Three hundred fifty-five patients showed one or more additional congenital craniofacial abnormalities (10%, Table 3). Most frequently, mandible abnormalities were observed (239 patients, 67%), which is consistent with the high frequency of Pierre Robin sequence. Four hundred forty-two patients displayed any congenital anomaly of other parts of the body (13%, Table 4), of which congenital anomalies of the circulatory system were the most frequent (32%). Patients with or without syndrome diagnosis were included in the above mentioned additional anomalies. Syndromic cases, cases with additional anomalies, and isolated cases were distinguished. Twenty-one percent of the common oral cleft patients were not isolated (10% CL/A, 13% CL/AP, 40% CP) (Table 5).
The Most Frequent Additional Congenital Abnormalities of the Head and Neck Area Besides Common Oral Clefts *

n = number of patients. Note that one patient may exhibit more than one abnormality.
Number of Patients With Congenital Anomalies of Other Parts of the Body *

n = number of patients. Note that one patient may have more than one affected body part.
Isolated Cases, Syndromic Cases, and Cases With Additional Anomalies Related to Category of Cleft *
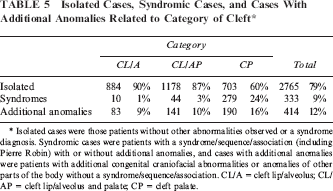
Isolated cases were those patients without other abnormalities observed or a syndrome diagnosis. Syndromic cases were patients with a syndrome/sequence/association (including Pierre Robin) with or without additional anomalies, and cases with additional anomalies were patients with additional congenital craniofacial abnormalities or anomalies of other parts of the body without a syndrome/sequence/association. CL/A = cleft lip/alveolus; CL/AP = cleft lip/alveolus and palate; CP = cleft palate.
One thousand one hundred nine patients (32%) were referred to a clinical geneticist, and a syndrome/sequence/association was observed in 212 patients (19% of the referred patients). Two thousand four hundred three patients were not referred to a clinical geneticist, and a syndrome/sequence/association was observed in 121 cases (5% of the nonreferred patients). In 1% of the CL/A patients, a syndrome diagnosis was established, and in 3% and 24% of the CL/AP and CP patients, respectively (all p < .001). The patients with a syndrome diagnosis were referred to a clinical geneticist in 20% of the CL/A patients, and in 39% and 37%, of the CL/AP and CP patients, respectively. Pierre Robin sequence was the most frequent diagnosis (Table 6), occurring only in CP patients, and CL/AP patients mostly showed trisomy 21 or trisomy 13.
The Most Frequent Syndromes/Sequences/Associations Observed in the Three Categories of Common Oral Clefts *

n = number of patients; CL/A = cleft lip/alveolus; CL/AP = cleft lip/alveolus and palate; CP = cleft palate.
Birth Prevalence
Birth prevalence rates were computed using the birth registry of the CBS and the birth dates of the patients. Between January 1, 1997, and December 31, 2006, there were 1,970,872 children born in The Netherlands. Because adopted/foster children most commonly are born outside The Netherlands, we decided to exclude adopted/foster children when calculating birth prevalence rates. Not every child with a common oral cleft who was born in 2006 had already been recorded in 2006. For instance, when a child was born in December 2006, it is readily possible that the cleft palate team had not been visited in 2006. Three thousand two hundred sixty-one patients with common oral clefts remained, resulting in a prevalence of 16.6 per 10,000 live births (1 per 604 live births). The birth prevalence rates between 1997 and 2006 are depicted in Figure 5.
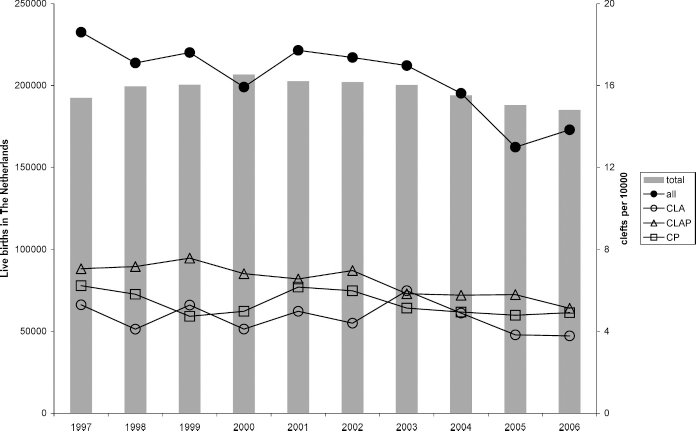
Birth prevalence rates of common oral clefts 1997 to 2006. The columns represent the number of live births in The Netherlands, and the quantity is shown on the left side. The dots and squares are the number of patients with a common oral cleft per 10,000 live births, and the right side shows the quantity.
Discussion
Recording and classification of common oral clefts should enable clinical and/or fundamental research. Among the objectives are surveillance for changes in frequencies of specific cleft types. Furthermore, clinical outcome largely depends on the phenotype of the cleft patient. Therefore, consistent description is required to evaluate any treatment strategy, to compare the results with other studies, and to improve interdisciplinary communication. This also applies for fundamental researchers who are searching for genetic factors responsible for different cleft types, gene functions, and gene-environmental interactions.
The NVSCA registration, as is shown in the results, can consistently describe all individual craniofacial abnormalities. All common oral clefts were divided into the three well-known categories: CL/A (28%), CL/AP (39%), and CP (33%). Other studies also have shown an excess of CL/AP (Magdalenic-Mestrovic and Bagatin, 2005; Gundlach and Maus, 2006; Meng et al., 2006). As is shown in several tables, many cleft types could be described within these categories. The first two examples of infrequent clefts cannot adequately be described by previous classifications without losing details about the individual abnormalities (Kernahan, 1971; Kriens, 1990; Smith et al., 1998; Koul, 2007; Liu et al., 2007). To assess the reliability of the acquired data, we have conducted a validation study (Rozendaal et al., 2010). Besides consistent description of all individual craniofacial abnormalities, the fast and easy recording is another strength of the NVSCA registration. This highly improves compliance by the physicians who actually assess cleft patients during their busy outpatient clinics. Any registration system has its flaws. Specific details about the cleft are not noted, such as the width of the cleft or the cleft proportion in incomplete cleft lip. Furthermore, the NVSCA registration is not an ongoing registration, and underestimation, for instance of congenital anomalies of other parts of the body, or other (discrete) craniofacial anomalies will result. It may be useful to record all cleft patients again after 6 years (van der Veen et al., 2006), thereby completing the additional abnormalities and enabling study of true isolated cleft cases. Additional anomalies of other body parts are also recorded. However, this part of the registration is based on verbatim descriptions of diagnosis, and incompleteness and inaccuracies result. Therefore, it could be beneficial to construct a recording form for each body part. We have introduced such a recording system for congenital anomalies of the upper limb (Luijsterburg et al., 2003), which is based on the same principles as the NVSCA registration. In this way, linkage between the different forms could allow more complete analysis, for instance of common oral clefts and upper limb anomalies. At last, this registry concerns live births, who live long enough to visit a cleft palate team. All still births as well as all live births who die before the first visit to a cleft palate team are not recorded, leading to an underestimation of birth prevalence rates.
In our study, the cleft palate teams assessed 91% of the patients with a common oral cleft within 12 months of age (85% within 5 months of age). This high percentage for early referral may be due to the adequate infrastructure and the sound embedding of the cleft palate teams in the Dutch welfare system.
Late referral (> 12 months of age, n = 299) may be partly due to the fact that the child is adopted. Fifty-five percent of the adopted children are seen for the first time in a cleft palate team later than 1 year of age. Most likely, these children are presented so late because they arrive later in life at the new parents or guest family. Of the nonadopted children, 6% visit the cleft palate team later than 1 year of age (221 patients). Another factor contributing to late referral may be children with cleft palate: 175 children are referred late (15% of all cleft palate patients). In 77 patients an (in)complete cleft of the hard and/or soft palate was observed (data not shown). The remaining 98 children showed submucous clefts and aplastic or hypoplastic hard and/or soft palates. Thus, the proportion of late referrals of CP children could be diminished by more thorough postnatal assessment by, for example, general physicians, midwives, gynecologists, and pediatricians.
Mean birth weight was the highest in the CL/A category. After exclusion of additional anomalies, no significant differences were observed. However, birth weight in all cleft patients was lower than in the general population (3453 g [3421 to 3485]) but the clinical explanation is obscure.
The male/female ratio was high in the CL/A group and the CL/AP group but was reversed in the CP group, as is in accordance with literature (Vallino-Napoli et al., 2004; Magdalenic-Mestrovic and Bagatin, 2005; Meng et al., 2006).
One in four patients showed a positive family history for any congenital abnormality and one in five for common oral clefts. Common oral clefts occurred less frequently among relatives in the CP group, 22% versus 26% CL/A and 29% CL/AP, respectively. Furthermore, in 9% a syndrome diagnosis/sequence/association was observed (one in four patients with a CP). In one in three patients, a clinical geneticist was consulted for further assessment. Only half of the patients with additional congenital abnormalities of the head and neck area and/or congenital anomalies of other parts of the body were referred to the clinical geneticist.
Ninety percent of the CL/A patients were isolated and 86% and 60% of the CL/AP and CP patients, respectively. Other studies have shown lower percentages of isolated cases (Tolarova and Cervenka, 1998; Stoll et al., 2000; Calzolari et al., 2004; Magdalenic-Mestrovic and Bagatin, 2005; Sarkozi et al., 2005; Calzolari et al., 2007; Harville et al., 2007). Methodologie factors that may cause variation in frequency of isolated cased have been discussed previously (Wyszynski et al., 2006): case definition and inclusion/exclusion criteria, timing of medical examination, variable clinical expression of associated anomalies, ability to establish syndrome diagnosis, patient selection, sample size, sources of ascertainment, and true population differences. Because the NVSCA registration is not an ongoing registration, it is likely to underreport additional anomalies. Furthermore, it is known that mild additional congenital abnormalities of the head and neck area are difficult to observe (Luijsterburg and Vermeij-Keers, personal communication), and that congenital anomalies of other parts of the body may reveal themselves later in life. As a consequence, relevant data are not recorded, leading to an underestimation of these additional abnormalities. This may account for the lower proportion of additional abnormalities and syndrome diagnoses. In our opinion, a clinical geneticist/pediatrician for an extensive assessment of the congenital anomalies should therefore see all cleft patients.
Common oral clefts are among the most frequent congenital anomalies. Our study revealed a prevalence of 16.6 per 10,000 live births over a 10-year period in The Netherlands. This is in accordance with prevalence rates in Western Europe (EUROCAT, 2007; IDCFA, 2007). Our birth prevalence rates are slightly underestimated because each year about 20 patients die in the first weeks of life, which is mostly before attending a cleft palate team (Anthony et al., 2005). All our recorded cases represent about 93% of the estimated live born cleft patients.
As the normal development of the primary and secondary palate evolves from 6 to 14 weeks of gestation, common oral clefts develop during the same period. As the different types develop at a specific time during this 2-month period, and additionally the underlying cell biological mechanisms are specific for different cleft types (Vermeij-Keers, 1990), a valid description of the cleft types is a condition sine qua non. Such a consistent description of common oral clefts is provided by the NVSCA registration. In this way, it may be possible to relate the observed cleft types to specific time periods, and subsequently, specific known and unknown genes that are expressed during these periods may be identified.
Footnotes
Acknowledgments.
We would like to express our gratitude to all cleft palate teams in The Netherlands and the NVSCA members and its board in particular. Without their enthusiasm and efforts, this system would not have succeeded. The cleft palate teams reside in Academic Medical Center in Amsterdam, Erasmus Medical Center-Sophia in Rotterdam, IJsselland Hospital in Capelle a/d IJssel, Leiden University Medical Center in Leiden/Juliana Hospital in The Hague, Medical Center Alkmaar in Alkmaar, Medical Center Leeuwarden in Leeuwarden, Rijnstate Hospital in Arnhem, Sophia Hospital in Zwolle, St. Elisabeth Hospital in Tilburg, University Medical Center Groningen in Groningen, University Medical Center Maastricht in Maastricht, University Medical Center St. Radboud in Nijmegen, University Medical Center Utrecht in Utrecht, Victor Veau Foundation in Almelo, and VU University Medical Center in Amsterdam.