
Abstract
Pfeiffer syndrome is a pleiotropic disorder characterized by multiple suture craniosynostosis, broad and medially deviated thumbs and great toes, and variable cutaneous syndactyly. We present the case of a 16-month-old boy with Pfeiffer syndrome type 2 who presented with intestinal malrotation for which the diagnosis was delayed. This is a rare complication of Pfeiffer syndrome, with few reported cases in the literature. This case illustrates the importance of recognizing gastrointestinal malrotation as a possible cause of feeding intolerance and persistent vomiting in patients with the severe forms of Pfeiffer syndrome.
Pfeiffer syndrome (MIM #101600) is an autosomal dominant condition characterized by craniosynostosis, midface hypoplasia, broad and medially deviated thumbs and great toes, variable cutaneous syndactyly, and brachydactyly. Three subtypes of Pfeiffer syndrome have been recognized based on the severity of the phenotype; although, there is significant clinical overlap (Cohen, 1993; Plomp et al., 1998; Robin et al., 1998; Oyamada et al., 2003; Oliveira et al., 2006). Pfeiffer syndrome type 1 is the most common type, usually associated with relatively mild manifestations including bicoronal craniosynostosis and broad medially deviated thumbs and halluces. Individuals with Pfeiffer syndrome type 1 usually have normal neurologic and intellectual development and a favorable prognosis. This is caused by mutations of either the FGFR1 or FGFR2 genes. On the other hand, Pfeiffer syndrome types 2 and 3 are characterized by more severe craniofacial and limb anomalies including contractures, syndactyly, central nervous system involvement, and poor prognosis and often result in early death. The main difference between these two types is the presence of a cloverleaf skull in Pfeiffer syndrome type 2, a feature that is absent in type 3. Both Pfeiffer syndrome types 2 and 3 are caused by mutations in the FGFR2 gene (Vogels and Fryns, 2006).
Visceral anomalies in Pfeiffer syndrome have rarely been reported. We report a case of a patient with Pfeiffer syndrome type 2 and intestinal malrotation, with particular emphasis on the importance of early recognition of this finding in patients with Pfeiffer syndrome who present with any symptoms of gastrointestinal disorder.
Case Report
The patient was the 3.375-kg (38.5th centile) product of a 39-week gestation cesarean section delivery to a 30-year-old gravida 2, para 1 woman and her 27-year-old unrelated husband. The parents were healthy and of normal intelligence. The mother was initially evaluated prenatally at 32 weeks' gestation for suspected craniosynostosis, which was confirmed by second-trimester fetal ultrasound. Fetal MRI was performed and showed significant deformity of the calvarium with trilobar skull configuration, acrocephaly, frontal bossing, hypertelorism, and bilateral proptosis. Other abnormalities identified included Chiari malformation type I, asymmetric lateral ventricles, and kyphosis at the cervicothoracic junction. Choanal stenosis was also a possibility although not clearly identified (Fig. 1). His stomach appeared normal in position, and there were no other apparent gastrointestinal tract anomalies. Amniocentesis reported a normal 46,XY karyotype, but genetic testing for craniosynostosis syndromes revealed a C.870G>C mutation in exon 8 (W290C) of the FGFR2 gene, consistent with the diagnosis of Pfeiffer syndrome type 2.
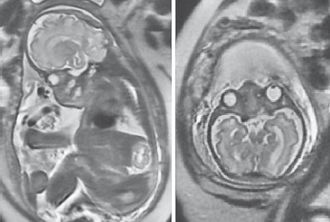
Fetal MRI images at 32 weeks' gestation. Note the significant deformity of the calvarium with trilobar skull configuration, frontal bossing, and hypertelorism. There is significant crowding at the craniocervical junction with some minimal extension of cerebellar tissue below the foramen magnum.
Postnatal brain MRI and head computed tomography confirmed the cloverleaf skull deformity with bilateral coronal and lambdoidal synostosis. He remained hospitalized after birth at an outside institution for 3 months, during which he underwent cranial surgery and had several other medical issues. He had cranial vault remodeling with extensive craniectomy and multiple suture craniotomies as well as tarsorrhaphy at age 2 weeks. A ventriculoperitoneal (VP) shunt was placed at 3 weeks of age for hydrocephalus, and he required a gastrostomy tube with Nissen fundoplication due to severe gastroesophageal reflux with no suspicion of gastrointestinal obstruction. A tracheostomy was required at age 6 weeks due to bilateral choanal stenosis.
His first genetic evaluation in our institution was at 3 Vi months of age while an inpatient, and he was seen several times during the following months (Fig. 2). In addition to the cloverleaf skull, he had contractures of the elbows with limited range of motion and typical broad thumbs and halluces, consistent with the diagnosis of Pfeiffer syndrome type 2. Over the following months he underwent craniofacial reconstruction with mandibular distraction osteogenesis at 8 months of age and posterior fossa decompression with suboccipital craniectomy at 16 months of age. He also was hospitalized multiple times during his first year of life, mostly related to respiratory issues and infections.
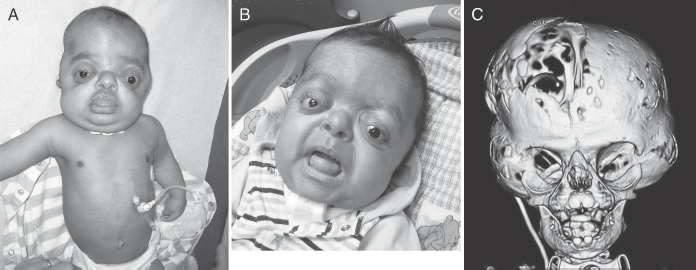
A: Patient at 5 months of age. B: Patient at 11 months of age. Note the brachy-turricephalic appearance of the head, hypertelorism, midface hypoplasia, and severe proptosis. C: Three-dimensional computed tomography reconstruction of the head at 6 months of age showing the typical cloverleaf skull.
The patient had chronic problems with feeding. He had persistent vomiting even after his posterior fossa decompression, raising questions regarding the efficacy of his Nissen procedure. At 16 months an upper gastrointestinal tract (GI) study performed did not show reflux; however, gastrointestinal malrotation was suggested after several reviews of the studies (Fig. 3). An upper endoscopy showed that the gastrostomy tube appeared to be blocking the pyloric outlet, and when exploring the second and third portions of the duodenum, the tip of the endoscope was noted by transillumination to be below the umbilicus in the midline, also suggestive of malrotation. The patient underwent exploratory laparotomy, at which time his malrotation was confirmed. The patient was noted to have his cecum in the left upper quadrant, and after eviscerating the small bowel, a very narrow mesentery and multiple Ladd bands across the duodenum were seen. After a difficult postoperatory period that was complicated with intra-abdominal fluid collection from a gastric leak at the gastrostomy site and an appendiceal stump leak, his emesis and feeding intolerance improved over the following days to the present date.
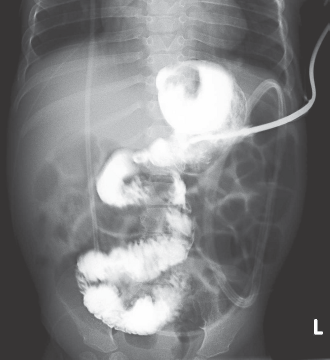
Upper GI at 16 months of age showing abnormal jejunal anatomy.
Discussion
We report a case of Pfeiffer syndrome type 2 with gastrointestinal obstruction caused by malrotation. The late recognition of the malrotation resulted in significant morbidity. There are few reports in the literature of visceral anomalies in craniosynostosis syndromes (Cohen and Kreiborg, 1993; Kodaka et al., 2004). In Apert syndrome, a similar craniofacial condition, gastrointestinal anomalies that include pyloric stenosis, esophageal atresia, partial biliary atresia with agenesis of the gallbladder, and imperforate or malposition of the anus were found in 1.5% of the patients (Cohen and Kreiborg, 1993). Pfeiffer syndrome types 2 and 3 have been reported with different gastrointestinal malformations including pyloric stenosis, hypoplastic gallbladder, absence of the lesser omentum, and anteriorly placed or imperforate anus (Hodach et al., 1975; Cohen, 1993; Ohashi et al., 1993; Tartaglia et al., 1997; Oyamada et al., 2003; Oliveira et al., 2006). However, intestinal malrotation has been reported once in both type 2 and type 3 Pfeiffer syndrome (Eaton et al., 1975; Barone et al., 1993).
Prenatal diagnosis of intestinal malrotation can be made using ultrasound and/or fetal MRI. A malpositioned stomach is usually the first sign, alone or in combination with other gastrointestinal tract anomalies that can be associated with malrotation such as bowel dilatation, volvulus, meconium peritonitis, cloacal malformation, or fetal ascites (Veyrac et al., 2004; Cassart et al., 2006; Biyyam et al., 2009). In the present case, the stomach was in normal position, and no other related anomalies on the prenatal images were suggestive of the diagnosis of intestinal malrotation.
Our patient had the typical features of Pfeiffer syndrome type 2. The severity of his phenotype, the presence of cloverleaf skull, and the mutation in the FGFR 2 gene were all consistent with this diagnosis. He had some gastrointestinal symptoms since early infancy, with gastroesophageal reflux and a feeding disorder that led to surgery for placement of a gastrostomy tube with a Nissen fundoplication. Of significance was his history of chronic feeding intolerance with no clear etiology. The integrity of his VP shunt was evaluated on multiple occasions because his symptoms were thought to be related to craniocervical compression, with no abnormalities having been found. He underwent posterior fossa decompression at 16 months of age. However, after this procedure his vomiting persisted, and it was not until then that other causes were evaluated, including the competency of his Nissen fundoplication and the potential for other anatomic causes. Even though the upper endoscopy found what could have been the only explanation for his persistent vomiting with the gastrostomy tube blocking the pyloric outlet, findings on the upper GI series led to the exploratory laparotomy and his final diagnosis.
This case illustrates that, although rare, gastrointestinal malrotation can be seen in patients with severe forms of Pfeiffer syndrome. The evaluation for this potentially lethal complication should be done in those patients with Pfeiffer syndrome who present with persistent feeding difficulties, particularly in those cases where other causes have been ruled out. This may result in preventing unnecessary and potentially harmful procedures as well as delineating the anatomic causes of vomiting and reflux before other interventions are performed, including VP shunt placements.