
Abstract
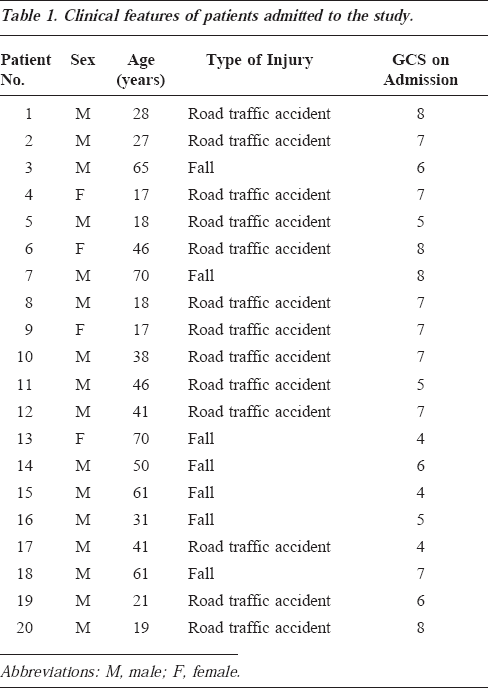
Background
On the basis of the contradiction between data on experimental head trauma showing oxidative stress-mediated cerebral tissue damage and failure of the majority of clinical trials using free radical scavenger drugs, we monitored the time-course changes of malondialdehyde (MDA, an index of cell lipid peroxidation), ascorbate, and dephosphorylated ATP catabolites in cerebrospinal fluid (CSF) of traumatic brain-injured patients.
Methods
CSF samples were obtained from 20 consecutive patients suffering from severe brain injury. All patients were comatose, with a Glasgow Coma Scale on admission of 6±1. The first CSF sample for each patient was collected within a mean value of 2.95 hours from trauma (SD=1.98), after the insertion of a ventriculostomy catheter for the continuous monitoring of intracranial pressure. During the next 48 hours, CSF was withdrawn from each patient once every 6 hours. All samples were analyzed by an ion-pairing high-performance liquid chromatographic method for the simultaneous determination of MDA, ascorbic acid, hypoxanthine, xanthine, uric acid, inosine, and adenosine.
Results
In comparison with values recorded in 10 herniated-lumbar-disk, noncerebral control patients, data showed that all CSF samples of brain-injured patients had high values (0.226 μmol/L; SD=0.196) of MDA (undetectable in samples of control patients) and decreased ascorbate levels (96.25 μmol/L; SD=31.74), already at the time of first withdrawal at the time of hospital admission. MDA was almost constant in the next two withdrawals and tended to decrease thereafter, although 48 hours after hospital admission, a mean level of 0.072 μmol/L CSF (SD=0.026) was still recorded. The ascorbate level was normalized 42 hours after hospital admission. Changes in the CSF values of ATP degradation products (oxypurines and nucleosides) suggested a dramatic alteration of neuronal energy metabolism after traumatic brain injury.
Conclusions
On the whole, these data demonstrate the early onset of oxygen radical-mediated oxidative stress, proposing a valid explanation for the failure of clinical trials based on the administration of oxygen free radical scavenger drugs and suggesting a possible rationale for testing the efficacy of lipid peroxidation “chain breakers” in future clinical trials.
Keywords
Introduction
Despite the implementation of many preventive measures, head injury remains the leading cause of death and disability in young adults in the Western world. Head injury management is primarily devoted to avoiding or limiting the development of secondary brain damage. Severe traumatic brain injury (TBI) is often characterized by diffuse cerebral tissue damage that involves different cerebral areas and areas far from the zone where the original impact occurred. 1 Because it is difficult to obtain specimens of posttraumatized human cerebral tissue for biochemical analyses, most molecular-level information on this pathological condition has been obtained from various animal models of TBI. Many of these studies have demonstrated that several pathophysiological mechanisms are involved in the onset of intracerebral complications, including release of excitatory amino acids, 2 changes of cell ionic permeability, 3 loss of mitochondrial function and consequent depression of energy metabolism, 4 increase of reactive oxygen species (ROS) generation, and subsequent irreversible modifications of several biologically important macromolecules. 5 Such numerous biochemical alterations of neuronal homeostasis are often worsened by phenomena of ischemia-hypoxia caused both by systemic and local problems of the correct cerebral tissue oxygenation.
However, using the “weight-drop” closed-head rodent trauma of mild-moderate gravity, which is characterized by diffuse axonal damage, as described by Marmarou et al, 6 we have very recently demonstrated that ROS-mediated lipid peroxidation is instantaneously triggered by trauma just at the time of impact (60 seconds after trauma), without the involvement of secondary phenomena of ischemia-hypoxia. We also found that that the decrease of cerebral energy metabolism takes place at successive times. 7 If this different temporal trend were to occur also in patients suffering from TBI, it might have relevance for determining the pharmacological strategy as a function of time after occurrence of TBI itself. In the last decade, several clinical studies with the specific purpose of improving the pharmacological therapies of TBI treatment have been carried out.8–11 Despite the use of different and promising drugs, most of the clinical trials did not produce positive results, particularly those that assessed the effectiveness of compounds with known in vitro efficacy toward ROS toxicity. 12 In response to such negative results, interesting explanations for the failure of pharmacological approaches have been presented. 13 It has even been suggested that ROS-associated cerebral tissue damage (such as lipid peroxidation), which occurs in different animal models of brain injury, including brain trauma, might be of minor clinical relevance. 14
Using biochemical analysis of the cerebrospinal fluid (CSF), this study was carried out to verify the time course of the eventual ROS-mediated oxidative stress (evaluated by determining malondialdehyde [MDA] and ascorbic acid levels) and of cerebral tissue alteration of the energy state (evaluated by monitoring variations of ATP catabolites) of comatose patients suffering from severe TBI (as measured by the Glasgow Coma Scale [GCS]) associated with diffuse cerebral damage. The biochemical and pharmacological significance of the present results are discussed.
Methods
Patient Population
This study was approved by the local Ethical Committee of Azienda Ospedaliera of Verona, Italy. Twenty patients with severe head trauma, admitted at any time from November 1998 to March 2000 to the Neurosurgical Intensive Care Unit, Department of Neurosurgery, University Hospital of Verona, and referred within 6 hours after trauma (mean, 2.95 hours; SD=1.98) were included in this study. The clinical features of patients admitted to this study are reported in Table 1. Seventy percent of the patients (14 of 20) were males, as might be expected with TBI, and the mean age was 39.25 years (SD=18.77; minimal and maximal ages, 17 and 65 years, respectively). Traffic accidents were responsible for head injury in the majority of cases. After stabilization, all patients were comatose with a mean GCS score (used for the evaluation of the clinical status) of 6±1. Patients were not included who were close to brain death on admission (GCS, 3; bilateral unreactive pupils) or who suffered from hypoxia (peripheral SatO2 <90) or hypotension (systolic arterial pressure <90 mm Hg), either before hospital care or at admission. In the neurosurgical emergency room, patients underwent evaluation of vital signs, neurological assessment, and computerized tomographic (CT) scanning. CT scans were classified according to the Traumatic Coma Data Bank. 15 Having a normal CT scan was an exclusion criterion. Within 6 hours after trauma, patients were subjected to ventriculostomy, performed using a closed-system ventricular catheter, preferably inserted into the right lateral ventricle. Each catheter was equipped with a fiberoptic pressure transducer on its tip and was secured to the skull by a bolt to allow simultaneous intracranial pressure monitoring and drainage (microventricular bolt 110-4MH, Camino, San Diego, Calif). Mean arterial blood pressure and cerebral perfusion pressure were also continuously monitored using a 7-channel recorder (Licox GMS Multimodality Monitoring, Kiel, Germany). Cerebral perfusion pressure was kept at 70 mm Hg or higher using mild hyperventilation (pCO2 level of 30-35 mm Hg), ventricular drainage, and a 2-mg/kg dose of mannitol. According to Marmarou et al, 16 patients who had readings of systolic blood pressure ranging from 90 to 120 mm Hg for at least 15 minutes during the CSF sampling period were considered hemodynamically unstable; therefore, to avoid a delayed superimposed secondary brain damage, which could have invalidated the understanding of the primary TBI, the patients were treated with intravenous administration of adrenaline to stabilize the systolic blood pressure and to, thus, allow their inclusion in the study. The outcome was assessed using the Glasgow Outcome Scale at 6 months follow-up. 17
Clinical features of patients admitted to the study.
Ten patients who did not suffer from any cerebral pathology, who were hospitalized for surgical reduction of a herniated lumbar disk, and from whom informed consent was obtained were subjected to lumbar CSF withdrawal immediately before surgical procedure. They represent the control group.
Csf Sampling
The collection of CSF samples was started immediately after catheter insertion in the operating room and repeated, when possible, every 6 hours for up to 48 hours. As an inclusion criterion, each patient had to undergo at least six of the nine CSF withdrawals. The CSF samples were centrifuged at 20,690 g for 10 minutes at 4°C, and the resulting supernatants were stored at -80°C until the treatment for biochemical analyses by high-performance liquid chromatography (HPLC). A daily check for meningitis or bacterial colonization was performed, and no occurrence of CSF infection was observed in any patient. At the end of the study, a total of 150 CSF samplings, including those of the control group, were carried out.
Preparation and Biochemical Analysis of CSF
The samples of CSF (500 μL) were deproteinized by adding a double volume of far-ultraviolet HPLC-grade acetonitrile and centrifuged at 20,690 g for 10 minutes at 4°C. The specimens were extracted twice with 2 mL of HPLC-grade chloroform. After each of the extractions, which were performed to remove acetonitrile, the specimens were centrifuged at 20,690 g for 5 minutes at 4°C. After the last centrifugation, the upper aqueous phases were gently collected, filtered through a 0.45-μm HV-Millipore filter (Millipore Corporation, Bredford, Mass) and analyzed by HPLC. Concentrations of MDA, ascorbic acid, hypoxanthine (Hyp), xanthine (Xan), uric acid, inosine (Ino) and adenosine (Ado) were analyzed on a 200-μL sample by an ion-pairing HPLC method 18 using a Kromasil 250x4.6 mm, 5-μm particle size column, with its guard column (Eka Chemicals AB, Bohus, Sweden), and using tetrabutylammonium hydroxide as the pairing reagent. Briefly, separation of different metabolites was obtained isocratically with a buffer having the following composition: 10 mM tetrabutylammonium hydroxide, 10 mM KH2 PO4, 0.25% methanol, pH 7.00. After each chromatographic run, the column was washed for 20 minutes with a second buffer, composed of 2.8 mM tetrabutylammonium hydroxide, 100 mM KH2 PO4, 30% methanol, pH 5.50, and then equilibrated with the separating buffer for at least 15 minutes before a new sample was loaded. The flow rate throughout the chromatographic runs was 1.2 mL/min, and the temperature was constantly kept at 23°C by thermostating the column with water-jacketed glassware.
The HPLC apparatus consisted of a Constametric 3500 dual pump system (ThermoQuest Italia, Rodano, Milano, Italy) connected with a SpectraSystem UV6000LP diode array detector (ThermoQuest Italia, Rodano), set between 200 and 300 nm of wavelength. Acquisition and analysis of the data were performed by a personal computer with the software package (ChromQuest) supplied by HPLC manufacturer. Areas, retention times, and absorbance spectra of different peaks of CSF sample chromatograms were compared with those of peaks of freshly prepared ultra pure standard chromatograms. This allowed us to both calculate concentrations of the various compounds at 267 nm of wavelength (the maximum of MDA absorbance spectrum) and identify with certainty the different metabolites.
Statistical Analysis
Differences between control and brain-injured patients, as well as time-course changes of the parameters under evaluation, were determined by the one-way analysis of variance, followed by the Newman-Keuls test; P <0.05 was considered significant.
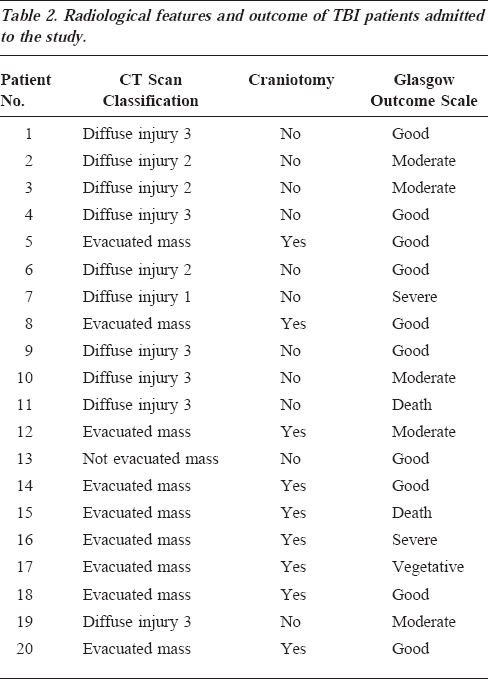
Results
Table 2 lists the selected clinical parameters of TBI patients admitted to this study. Regarding the type of lesions observed at the CT scan, 10 were classified as “diffuse injury” and 10 as “focal lesions” (nine of them evacuated). Craniotomy was performed in nine of 20 patients. In the survivors, the mean follow-up evaluated by the Glasgow Outcome Scale was 6±1 months (minimal and maximal values, 3 and 11 months, respectively); 16 patients had a favorable outcome (from good to moderate) and four an unfavorable outcome (one had a severe disability, one was in a vegetative state, two died 90-190 hours after hospital admission). Mean values of intracranial pressure, cerebral perfusion pressure, and mean arterial blood pressure of each patient are shown in Figure 1 and indicate that the threshold of cerebral perfusion pressure >80 mm Hg was achieved in the majority of patients. The mean time of monitoring duration was 156 hours (range, 48-341 hours).
Radiological features and outcome of TBI patients admitted to the study.
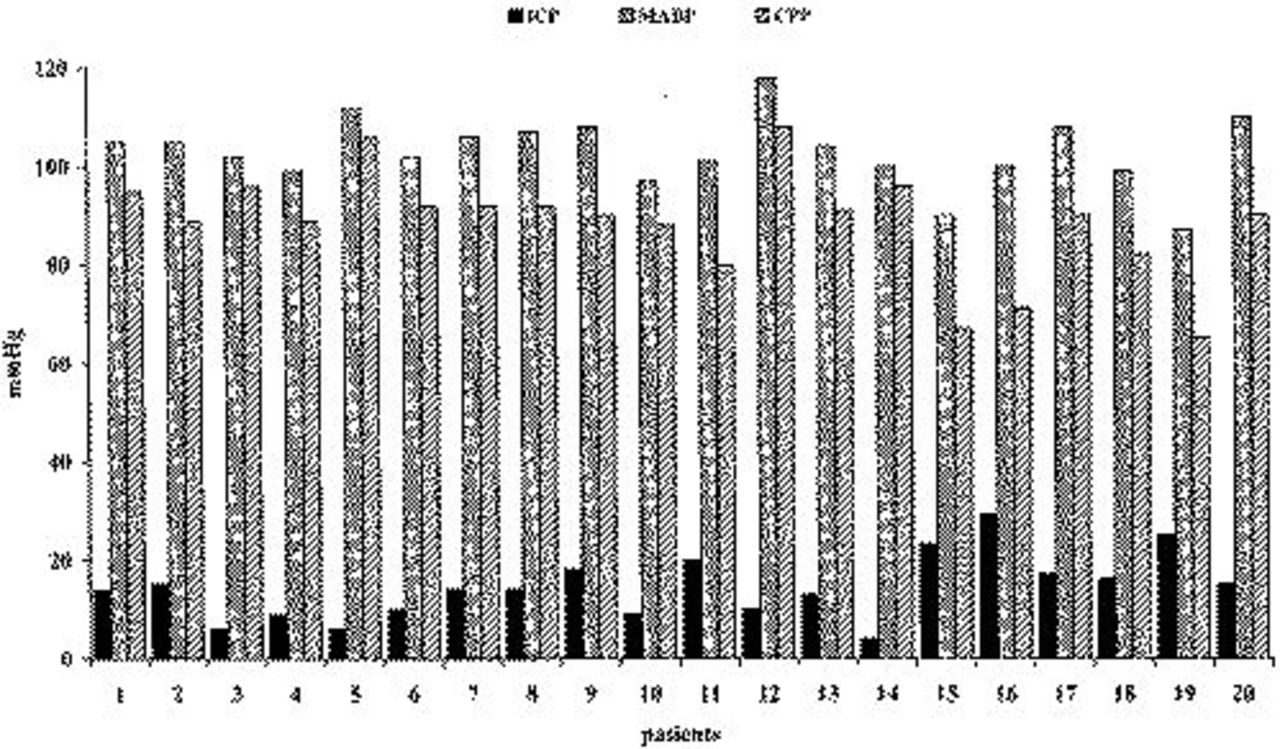
Mean values of intracranial pressure (ICP), mean arterial blood pressure (MABP), and cerebral perfusion pressure (CPP) of 20 TBI patients calculated from values recorded during 48 hours of continuous monitoring started at the time of their hospital admission. SDs are omitted for the sake of clarity.
Figure 2 lists time-course changes of MDA determined in the CSF of head-injured patients, starting within 6 hours after trauma occurrence. At the zero time point (corresponding to the first CSF withdrawal effected in the operative room 2.95 hours after head injury, SD=1.98), MDA had a concentration of 0.226 μmol/L of CSF (SD=0.196), which remained practically constant for the subsequent 12 hours. Values of this biochemical index of lipid peroxidation slowly decreased thereafter, although 48 hours after hospital admission, detectable MDA concentration in the CSF was still present (0.072 μmol/L of CSF; SD=0.026). Of note, MDA in the CSF of the control group, which included patients who were subjected to surgical intervention of herniated lumbar disk and who did not suffer from any cerebral pathology, was undetectable. The existence of oxidative stress associated with head trauma was clearly confirmed by changes in CSF ascorbate values, reported in Figure 3. In fact, the concentration of the most abundant water-soluble cerebral antioxidant was 151.66 μmol/L of CSF (SD=28.99) in the group of control patients, whereas in TBI patients, at the zero time point a value of 96.25 μmol/L of CSF (SD=31.74) was recorded. Hence, a significant CSF ascorbate decrease of 36.5% was produced within 2.95 hours (SD=1.98) after the trauma occurred (P <0.001). Ascorbate oxidation proceeded for the next 12 hours and, at this point, its minimal value was determined (79.79 μmol/L of CSF; SD=24.58; -47.4%; P <0.001, with respect to the value of the control group). The successive CSF withdrawals showed that the ascorbate level tended to restore slowly, even though significantly lower ascorbate values were observed at up to 36 hours after patient hospitalization (94.02 μmol/L of CSF; SD=21.87; -38.0%; P <0.001, compared with the control group). Only after 48 hours from the beginning of withdrawals, ascorbate levels in the CSF of TBI patients were comparable to those recorded in the control patients (120.40 μmol/L of CSF; SD=39.88).
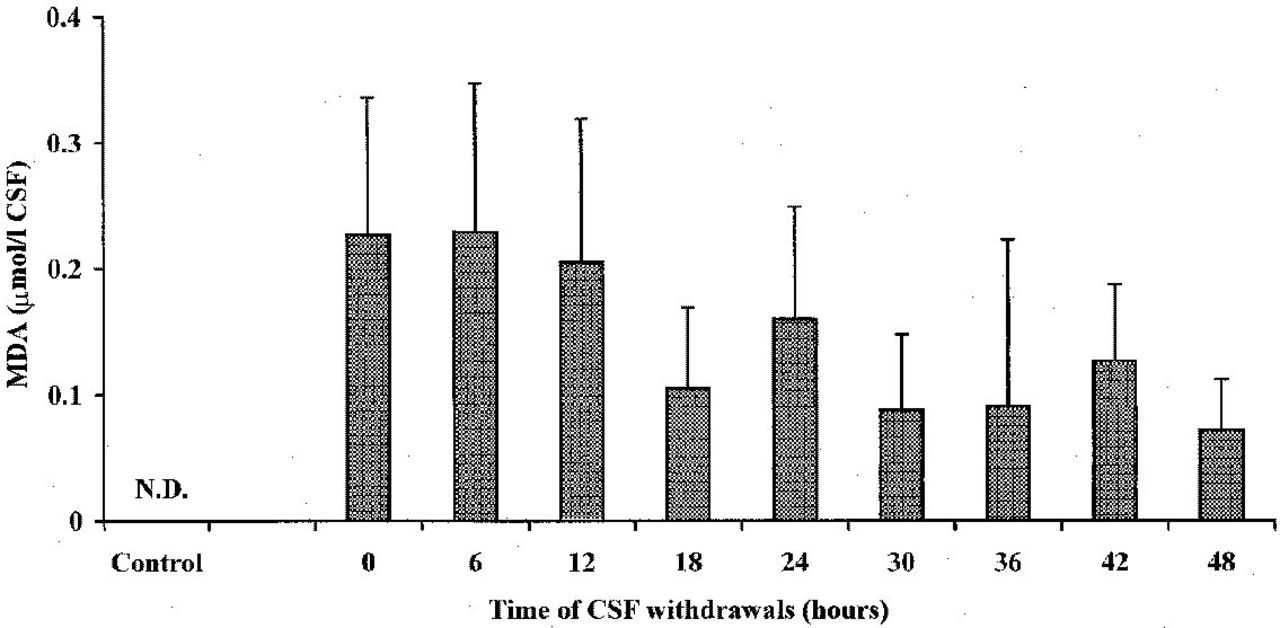
Time-course changes of MDA concentration in the CSF of 20 TBI patients. The control group was represented by 10 herniated lumbar disk patients not suffering from any cerebral pathology. Columns are the mean of 13 to 20 values. SDs are indicated by vertical bars. Zero time corresponds to the first CSF withdrawal performed a mean value of 2.95 hours (SD=1.98) after trauma occurred. All MDA values of TBI patients were significantly higher than those of control patients (P<0.001). Abbreviations: ND, not detectable.
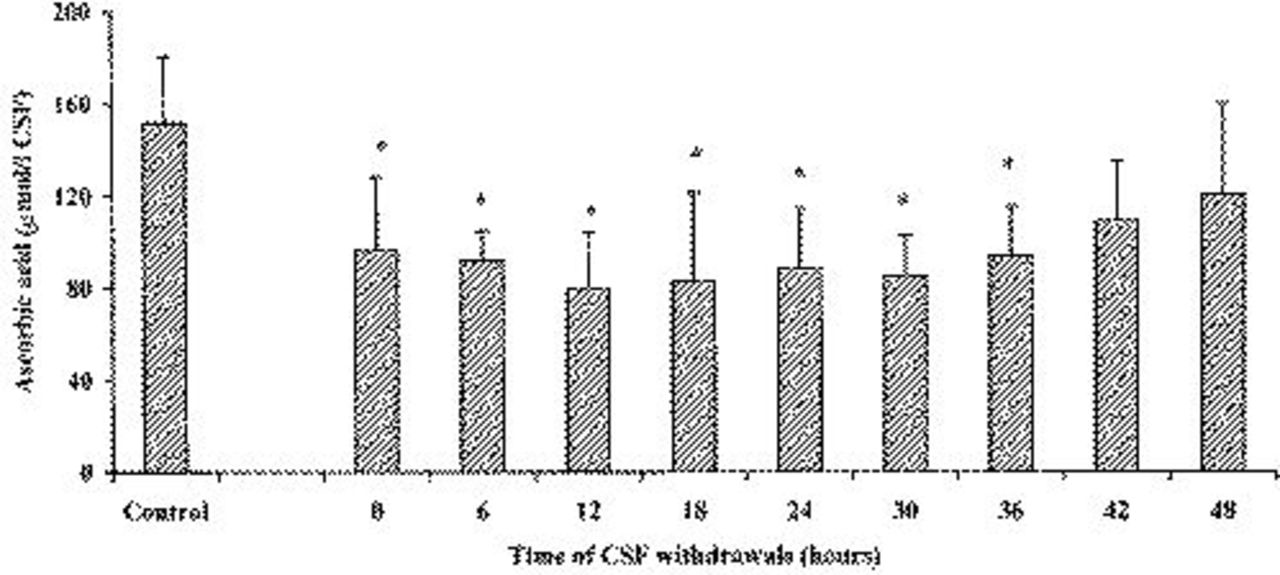
Variations of ascorbic acid concentrations in the CSF of 20 subjects after TBI. The control group was represented by 10 herniated lumbar disk patients not suffering from any cerebral pathology. Columns are the mean of 13 to 20 values. SDs are indicated by vertical bars. Zero time corresponds to the first CSF withdrawal performed a mean value of 2.95 hours (SD=1.98) after trauma occurred.
Figure 4 reports variations of Ino and Ado in the CSF of patients after TBI. Both nucleosides showed significantly higher values at the time of the first CSF withdrawal in patients suffering from TBI, when compared with corresponding values determined in herniated patients with no cerebral pathologies. This should indicate the onset of energy derangement, ie, imbalance between ATP production and consumption, within the lag time existing between TBI occurrence and the time of hospital admission. We observed a tendency in TBI patients for Ino and Ado values to return to normal levels 18 to 24 hours after hospital admission, even though levels of both nucleosides in TBI patients were similar to those of the control group only 48 hours after the first CSF withdrawal.
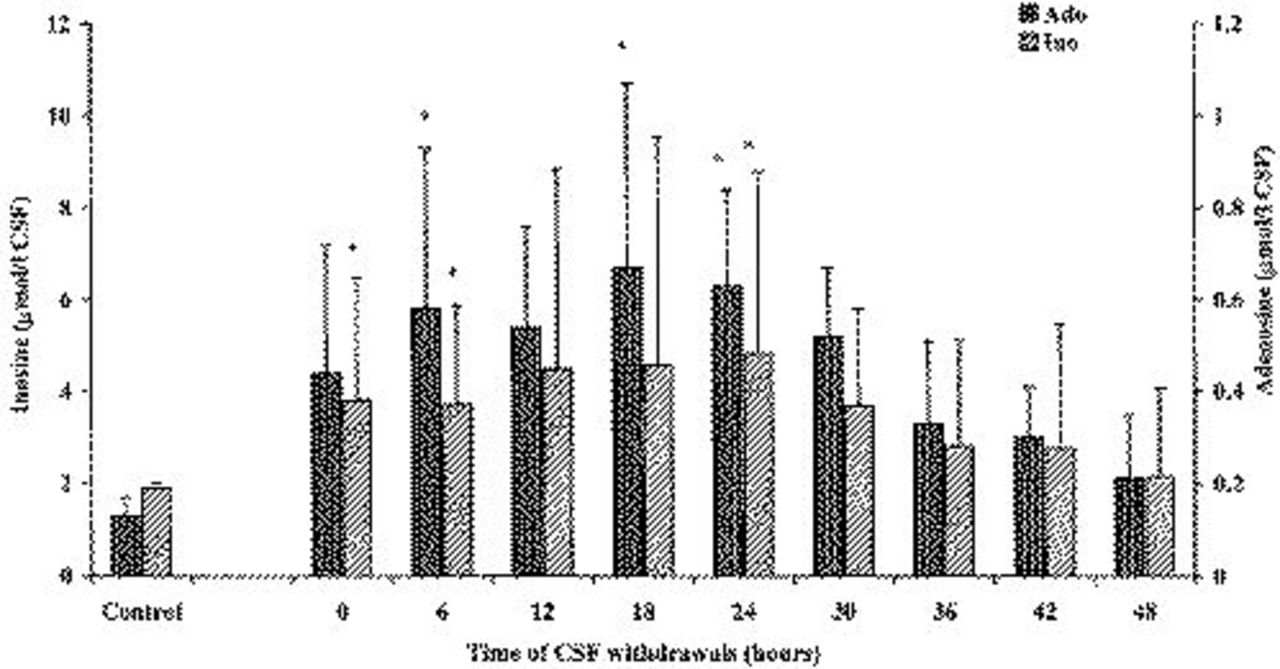
Concentration of nucleosides (Ino and Ado) in the CSF of patients after TBI. The control group was represented by 10 herniated lumbar disk patients not suffering from any cerebral pathology. Columns are the mean of 13 to 20 values. SDs are indicated by vertical bars. Zero time corresponds to the first CSF withdrawal performed a mean value of 2.95 hours (SD=1.98) after trauma occurred.
In Figure 5, time-course variations of CSF concentrations of Hyp, Xan, and uric acid in TBI patients and in the control group are reported. Compared with values recorded in control group subjects, Hyp and Xan values in TBI patients increased remarkably (+625% and +930%, respectively) after the first CSF sampling. Both oxypurines reached their maximal levels 12 hours after the beginning of CSF withdrawal, when Hyp was 8.58-fold higher and Xan was 11.48-fold higher than corresponding values determined in control patients (P <0.001). A progressive decrease of CSF concentrations of these compounds deriving from ATP catabolism was observed thereafter, although in the last CSF withdrawal, Hyp was 3.26-fold higher and Xan was 6.32-fold higher than the corresponding values recorded in the control group. Uric acid, ie, the most oxidized of natural oxypurines, had a different time course with respect to its precursors Hyp and Xan. In fact, although the mean uric acid values in CSF of TBI patients were higher than the concentrations recorded in the control group, the high dispersion of data did not allow to reach statistical significance at any time point.
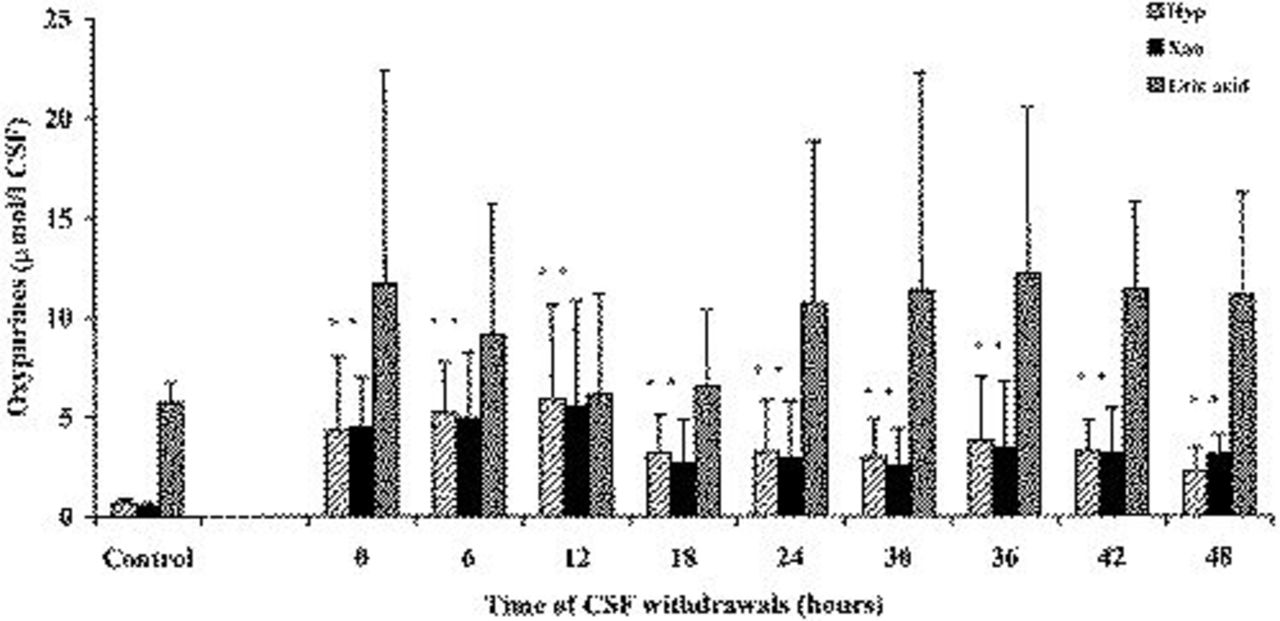
Concentration of oxypurines (Hyp, Xan, and uric acid) in the CSF of patients after TBI. The control group was represented by 10 herniated lumbar disk patients not suffering from any cerebral pathology. Columns are the mean of 13 to 20 values. SDs are indicated by vertical bars. Zero time corresponds to the first CSF withdrawal performed a mean value of 2.95 hours (SD=1.98) after trauma occurred.
Discussion
In the past few decades, the involvement of ROS in contributing to cell and tissue damage associated with several human pathologies of great clinical relevance has been demonstrated widely. Through the evaluation of different biochemical indices representative of ROS-induced irreversible modification of biologically fundamental macromolecules, it has been shown that chronic inflammations, 19 diabetes, 20 viral infections, 21 tissue ischemia, and reperfusion22–24 are characterized by ROS production and consequent decrease of cell antioxidant defenses. It has also been confirmed that in several neurological disorders, such as Alzheimer disease, 25 Parkinson disease, 26 multiple sclerosis, 27 etc, ROS overproduction is responsible for causing different types of cell injury.
Notwithstanding the fact that different experimental models of TBI have shown the occurrence of ROS-mediated cell damage, such as the induction of a lipid peroxidation reaction chain,5,7,28 clinical trials conducted with several promising drugs, capable of scavenging or inhibiting ROS, did not produce significant amelioration of TBI patient outcome (prognosis).11–13,29 Therefore, ROS generation in human beings after TBI still needs to be proven clearly, even if a recent indication of oxidative stress occurrence in head-injured patients has indirectly been obtained. 30 Unfortunately, these data suffer from the limitation that lipid peroxidation was determined in plasma rather than in the CSF; because plasma does not reflect cerebral metabolic changes only, it is hard to affirm that the increase in plasma lipid peroxidation was unequivocally a result of oxidative stress-mediated brain damage after TBI.
Data reported in the present study demonstrated that ROS-mediated oxidative stress occurs in patients suffering from severe TBI and, most importantly for possible pharmacological strategies, it is triggered in the early phase after TBI itself. In fact, our group of comatose TBI patients, although limited in number (n=20), had a mean MDA value at the time of the first CSF sampling (effected after a mean value of 2.95 hours, SD=1.98, after TBI occurrence) of 0.226 μmol/L of CSF, whereas this value, which is indicative of ROS-induced lipid peroxidation, was undetectable in control patients. The subsequent MDA determinations, accomplished every 6 hours for up to 48 hours after hospital admission, showed a slow but constant decrease of concentration in the CSF of TBI patients. This implies that the initiation and propagation of the lipid peroxidation reaction chain, induced by massive ROS production after TBI, takes place in a short time interval after head trauma. Therefore, this confirms the recent experimental evidence, observed in rats subjected to the rodent closed-head trauma model according to Vagnozzi et al, 7 which shows the induction of lipid peroxidation only 60 seconds after mild TBI. In this animal model, characterized by diffuse brain injury, we found a maximum of cerebral MDA generation within 2 hours after trauma. 7 The aforementioned observation and the results obtained in vitro, showing a lag time of 30-60 minutes for the propagation phase of ROS-induced lipid peroxidation, 31 corroborate the data reported in the present study, which indicate an early onset of oxidative stress after TBI. The consequent cerebral tissue lipid peroxidation takes place in the time interval from trauma occurrence to first CSF sampling (the mean value of which in our group of patients was 2.95 hours, SD=1.98), performed immediately after hospital admission. Products that derive from the degradation of peroxidized polyunsaturated fatty acids of membrane phospholipids, including MDA, are initially released in the extracellular space and then flow together in CSF, thanks to the washout activity of CSF itself. The final result is the presence of detectable MDA in liquor of TBI patients. In this light, we have recently demonstrated the relative facility of MDA to cross the cell membrane during hydrogen peroxide-induced erythrocyte lipid peroxidation, so that most of MDA produced under these experimental conditions was detected in the extracellular milieu. 32 It is also worth recalling that the onset of oxidative stress in the time interval from the beginning of symptoms to hospital admission (3.8 hours) was previously reported to have occurred also in patients suffering from acute myocardial infarction, in which elevated plasma MDA with respect to controls (healthy subjects and noncardiac patients) was recorded at the time of hospital admission. 33 The depletion of CSF ascorbate, already observable in the first CSF withdrawal, strongly supports the evidence that massive ROS production is generated shortly after trauma. Such a remarkable decrease of the most abundant water-soluble brain antioxidant certainly compromised cerebral ROS scavenging defenses. Moreover, because ascorbate is strictly related to the redox cycle of vitamin E (the main membrane lipid-soluble antioxidant capable of breaking the lipid peroxidation reaction chain), a close relationship between MDA generation, ascorbate decrement in CSF, and induction of oxidative stress in cerebral tissue after TBI is possible. It may also be hypothesized that the presence of detectable MDA levels in CSF 48 hours after TBI, in spite of quasi-normal CSF ascorbate concentrations, is mainly a result of the slow kinetics of MDA elimination caused by the ability of this bifunctional aldehyde to cross-react with amino acids, proteins, and sugars, rather than with a persistent lipid peroxidation. The existence of energy metabolism imbalance in cerebral tissue of patients suffering from TBI was clearly evidenced by the remarkable increase in CSF of dephosphorylated compounds, such as Ino, Ado, Hyp, Xan, and uric acid, deriving from ATP catabolism. This finding, already evidenced in the cerebral tissue of rats subjected to the impact acceleration model of closed-head trauma, 7 suggests that the mitochondrial dysfunction leading to alteration of the cell energy state might be one possible source of ROS generation resulting from the loss of efficiency to fully manage the tetravalent reduction of molecular oxygen into water. 34
Notwithstanding the relatively limited number of patients, which did not allow us to find a correlation between patient outcome and level of the biochemical parameters representative of ROS-mediated oxidative stress, it might be affirmed that our findings help explain the possible reasons for failure of the ROS scavenger pharmacological therapy of TBI.11–13,29 In fact, our data indicate that the administration of ROS scavenger or ROS inhibiting drugs could be almost completely ineffective unless given to TBI patients immediately after trauma (ie, within a few minutes). On the other hand, treatments with ROS scavengers effected hours after trauma, as it occurs in the majority of cases, might be successful only when considering the administration of lipid peroxidation chain breakers (such as vitamin E), eg, only if the drug administered is capable of interrupting the propagation phase of lipid peroxidation. Treatments with other types of drugs, exclusively or mostly acting as direct ROS scavengers,11,12 should be ineffective because massive ROS production seems to occur much earlier than any possible drug administration.5,7
In conclusion, we have demonstrated in the present study the occurrence of early onset of lipid peroxidation in patients suffering from TBI. This finding might help to make the choice of antioxidant drug and its administration protocols easier, with the aim of optimizing results in terms of patient outcome. Further studies to correlate clinical and biochemical parameters are in progress.
Footnotes
Acknowledgments
We thank the nurses of the Neurosurgical Intensive Care Unit, Department of Neurosurgery, of the University Hospital of Verona, whose efforts were essential for the accomplishment of this study.