
Abstract
Introduction
One of the major advances in liver research in the past decade was the ability to isolate distinct liver cell populations. Although there are established methods of isolating hepatocytes, cholangiocytes, and stellate cells, before this study no technique for liver fibroblast isolation had been devised. Consequently, we developed a technique to isolate primary rat liver fibroblasts.
Methods
Fibroblasts were isolated from a freshly perfused rat liver with a modification of the procedure for isolation of rat cholangiocytes. Cell markers were assessed with the use of confocal immunofluorescence. Cell morphology was assessed with transmission electron microscopy. Expression of procollagen-1 was assessed by reverse transcription polymerase chain reaction.
Results
The appearance of cells with fibroblast morphology was first noted at 48 hours, and almost all cells in culture had fibroblast morphology at 96 hours. Putative fibroblasts stained for vimentin, but not for smooth muscle actin, von Willebrand factor, or cytokeratins. Cell morphology was consistent with that of fibroblasts and showed no features of epithelial, endothelial, or smooth muscle cells. Liver fibroblasts expressed procollagen-1 mRNA.
Conclusion
Primary isolated rat fibroblasts can be produced from a freshly perfused rat liver with a modification of standard cell culture methods. The role of fibroblasts in liver physiology can now be studied directly.
Introduction
Progress in the study of liver physiology and pathophysiology has advanced significantly during the past decade. One of the major advances in liver research in this period has been the ability to isolate cell populations within the liver other than hepatocytes. 1 As a group, these nonhepatocyte liver cell populations have been referred to as nonparenchymal cells (NPCs). Techniques for the isolation of bile duct epithelia have added to the understanding of diseases that primarily affect these cells,2,3 and techniques for the isolation of hepatic stellate cells have added to the understanding of hepatic fibrosis. 4 Isolation of separate NPC populations has traditionally been performed using a variety of biochemical techniques, such as differential centrifugation and immunological approaches. 1 NPCs have been used in short- and long-term cultures for both biochemical and functional studies.
Although methods for the isolation of NPCs, such as bile duct epithelia, Kupfer cells, and hepatic stellate cells, have been established, other NPC populations such as hematologic, neural, and fibroblastic cells have not been isolated successfully. In this article, we describe a method of isolating primary hepatic fibroblasts using a modification of the method of isolating other NPCs. We demonstrate that the cells isolated are in fact fibroblasts because of their morphologic appearance and the absence of cell markers typical of other cell lineages, and also because they express procollagen, which demonstrates that they are capable of matrix synthesis.
Materials and Methods
Materials
Minimum essential medium, Dulbecco modified Eagle F-12 medium, Roswell Park Memorial Institute (RPMI) medium, penicillin-streptomycin, gentamicin, Fungizone (Bristol-Myers Squibb, Princeton, NJ), and fetal calf serum were obtained from Gibco BRL (Carlsbad, Calif). Bovine serum albumin, collagenase, hyaluronidase, and deoxyribonuclease I, were obtained from Sigma Chemical Co. (St. Louis, Mo). Pronase protease was obtained from Calbiochem (San Diego, Calif). We obtained 100-μ pore mesh from Selfar America (Kansas City, Mo). All other chemicals were of the highest quality commercially available.
Animals
Adult male Sprague-Dawley rats weighing from 180 to 300 g were used for all studies. The animals were anesthetized with pentobarbital sodium (50 mg/kg) before all studies, and they were killed by hemorrhage before they regained consciousness. Animal treatment was performed in accordance with the Yale Animal Care and Use Committee guidelines.
Collagenase Perfusion of the Liver
The NPCs to be isolated were obtained from the hepatic hilum using methods previously described. 1 Each rat was anesthetized as described in the previous section and placed supine on a dissecting tray. A midabdominal incision was made so that the liver was completely exposed. The portal vein was cannulated with a 16-gauge catheter, and the inferior vena cava was ligated distally to allow drainage of perfusate. The liver was perfused at 40 mL/min with Hanks’ A solution (Invitrogen Corp., Carlsbad, Calif) that contained NaCl (120 mM), KCl (5 mM), KH2PO4 (0.4 mM), Na2HPO4 (0.2 mM), NaHCO3 (25 mM), ethyleneglycol tetra-acetic acid (0.5 mM), and d-glucose (0.1% w/v) for 91/2 minutes. At this point, blanching of the liver was evident. The liver was then perfused at 40 mL/min with Hanks’ B solution (Invitrogen Corp.) that contained NaCl (120 mM), KCl (5 mM), KH2PO4 (0.4 mM), Na2HPO4 (0.2 mM), NaHCO3 (25 mM), MgSO4 (0.4 mM), MgCl2 (0.5 mM), CaCl2 (3 mM), and d-glucose (0.1%) that contained collagenase (300 mg/L) for approximately 20 minutes, until liver softening was evident. The liver was placed in a 10-cm tissue culture dish with cold Leibovitz/s L-15 medium (Invitrogen Corp.) that contained 1.26 mM CaCl2, 5.3 mM KCl, 0.441 mM KH2PO4, 0.986 mM MgCl2, 0.814 mM MgSO4, 138 mM NaCl, 1.34 mM Na2HPO4, 5 mM d-galactose, 5 mM Na pyruvate, 0.025 phenol red, and various amino acids and vitamins, then rinsed and placed in a new culture dish with fresh Leibovitz/s L-15 medium. The hepatic hilum was removed manually from peripheral hepatocytes and transferred to a fresh dish for NPC isolation.
Nonparenchymal Cell Isolation and Fibroblast Differentiation
The hepatic hilum was manually minced in the presence of pronase solution. The minced tissue was placed in a flask and shaken for 30 minutes at 37°C. This suspension was then passed through a mesh with 100-μ pores and spun at 1600 rpm for 5 minutes. Tissue remaining on the mesh was placed in hyaluronidase solution and shaken again for 30 minutes at 37°C. Cells were resuspended in ice-cold RPMI medium that contained 0.006% deoxyribonuclease I. Cells were then washed twice in RPMI medium and then resuspended in plating medium (Dulbecco modified Eagle F-12 medium that contained 2% penicillin-streptomycin, 3% fetal calf serum, 0.3% gentamicin, and 0.1% Fungizone). Freshly isolated NPCs was placed in plating medium as described in the section above in individual wells of a 24-well plate at 37°C. Cells were examined after 6, 48, and 96 hours to observe cell morphology.
Confocal Immunofluorescence
Confocal immunofluorescence was performed on hepatic fibroblasts using either indirect (von Willebrand factor, smooth muscle actin) or direct (vimentin, cytokeratin) immunofluorescence. Positive controls were performed on either 10-μ sections of rat liver (von Willebrand factor, cytokeratin) or primary rat myofibroblasts (α-smooth muscle actin). Ninety-six-hour cultures of primary hepatic fibroblasts grown on glass coverslips were washed twice in phosphate-buffered saline (PBS). Cells were fixed in ice-cold methanol for 10 minutes, rinsed twice quickly with ice-cold acetone, and washed twice with PBS for 30 minutes. For direct immunofluorescence experiments, cells were placed in PBS that contained 1% bovine serum albumin and either mouse monoclonal antivimentin Cy3 conjugate (Sigma Chemical Co.) at 1:50 or mouse monoclonal anti-pan cytokeratin fluorescein isothiocyanate conjugate (Sigma Chemical Co.) at 1:50 for 60 minutes at room temperature. Cells were washed with PBS at room temperature three times, covered with aqueous mounting medium (Molecular Probes, Eugene, Ore), and affixed to glass slides. For indirect immunofluorescence experiments, cells were fixed and prewashed as before. Cells were exposed to either rabbit anti-von Willebrand factor (Sigma Chemical Co.) at 1:800 or mouse monoclonal anti-α smooth muscle actin (Sigma Chemical Co.) at 1:25 in PBS that contained 1% bovine serum albumin for 60 minutes at room temperature. Cells were then washed three times with PBS at room temperature and labeled with either goat antirabbit Alexa 488 conjugate (Molecular Probes) at 1:400 (von Willebrand factor) or goat antimouse Cy3 conjugate (Jackson Immunoresearch Laboratories, West Grove, Pa) at 1:3000 (smooth muscle actin). Postlabeling washing and mounting of cells was performed as described above.
In separate experiments, immunofluorescence using the same methods was performed on 10-μ sections of the hepatic hilum obtained after collagenase removal of hepatocytes, as described above. Confocal imaging of fixed cells or liver sections was performed using a Zeiss LSM 510 confocal imaging system (Carl Zeiss Co., Oberkochen, Germany). Cells were excited with a krypton/argon laser at either 488 nm (for Alexa 488 and fluorescein isothiocyanate conjugates) or 568 nm (for Cy3 conjugates). Images were observed at >515 nm (for Alexa 488 and fluorescein isothiocyanate conjugates) or >585 nm (for Cy3 conjugates). In all experiments, the experimental specimens were observed using settings identical to those used with positive control specimens, so that a threshold level of “positive” fluorescence was defined on the basis of positive control experiments. These settings were used for each experiment within a set of antibody conditions.
Transmission Electron Microscopy
Transmission electron micrographs were obtained from centrifuged aggregates of hepatic fibroblasts. Ninety-six-hour cultures of primary hepatic fibroblasts were detached from their plates using trypsin and centrifuged for 5 minutes at 1000 rpm. The aggregates were fixed overnight in 0.1 M sodium cacodylate that contained 3% glutaraldehyde. Cells were postfixed in OsO4, dehydrated in ethanol, and embedded in Epox 812 embedding resin (Ernest F. Fullam, Latham, NY). Samples were sectioned and stained with uranyl acetate and lead citrate. Electron micrographs were collected using a Philips 300 electron microscope (Philips, Eindhoven, The Netherlands) and photographed using standard photographic techniques.
Reverse Transcription Polymerase Chain Reaction
Expression of procollagen-1 mRNA in various cell types was examined with the use of reverse transcription polymerase chain reaction (RT-PCR). Total RNA was synthesized from primary fibroblasts, myofibroblasts, and bile duct epithelia using chaotropic methods (RNAwiz; Ambion, Austin, Tex). Forward and reverse oligonucleotide primers were designed using the GenBank published sequence of rat procollagen-1. The primers are 5′-GCTCAGTATTCTGACAAAGGAGT-3′ and 5′-ATTTGCTCCAGGGTTGCAG-3′. After the RNA was treated with DNase to remove contaminating genomic DNA, cDNA was synthesized using sup reverse transcriptase (Gibco Life Technologies, Carlsbad, Calif) and random hexamer primers. cDNA was used as a template to amplify a predicted 650-base pair PCR product with the above-described procollagen-1 primers according to the following protocol: 94°C for 5 minutes; 30 cycles of 94°C for 30 seconds, then 62°C for 1 minute, then 72°C for 1 minute; and 72°C for 10 minutes. PCR products were run out on a 1.5% agarose-Tris-acetate-ethylenediamine tetra-acetic acid gel, stained with ethidium bromide, and photographed under ultraviolet transillumination.
Results
Differentiation of Primary Hepatic Fibroblasts over Time
Primary rat fibroblasts differentiated from NPCs during the course of several days. As seen in Figure 1, within 48 hours, cells with spindle morphology became evident (30–60% of cells observed), and within 96 hours, 98% of cells showed spindle morphology. In five preparations, only two cells retained a rounded morphology. This development demonstrates that, by using the methods described above, we were able to isolate cells with morphologic characteristics typical of fibroblasts from a freshly perfused rat liver. These cells represent a population of primary cells selected by the culture conditions used in isolation.
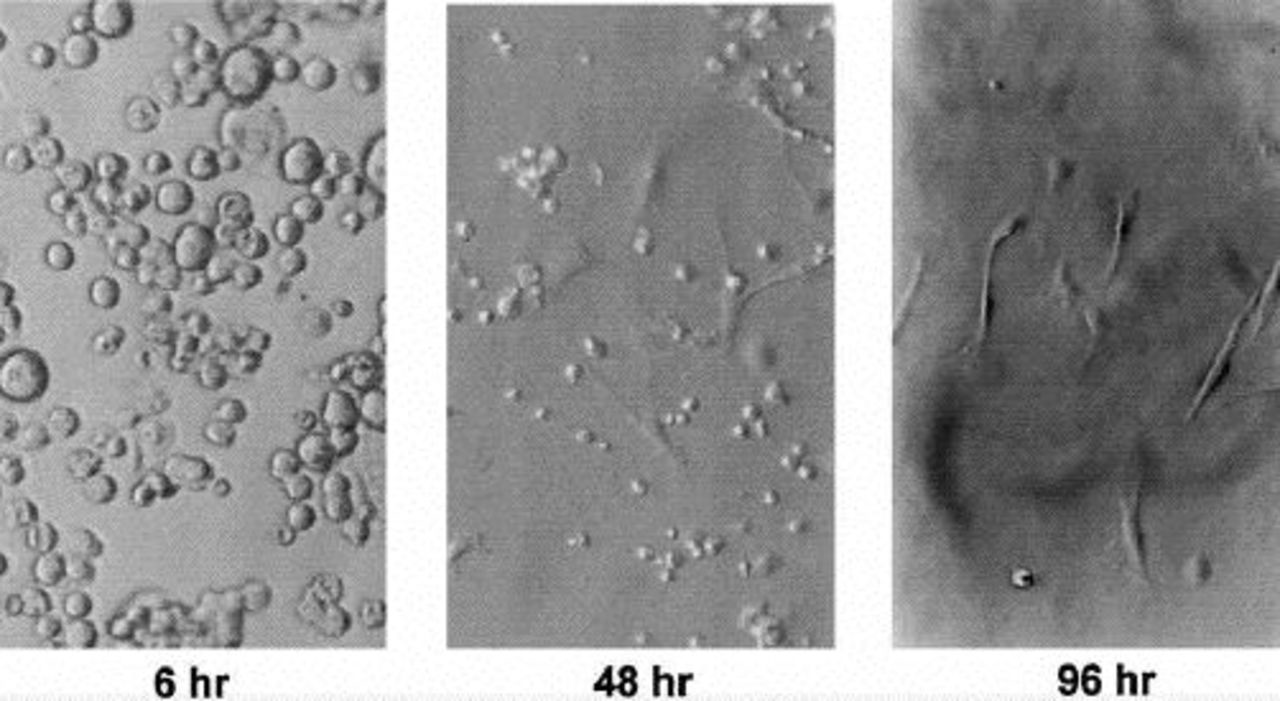
Nonparenchymal cells in culture assume spindle morphology after 48 to 96 hours. Rat NPCs were isolated from freshly perfused rat livers as described. NPCs were placed in culture medium and serially observed at 6, 48, and 96 hours. At 6 hours, all cells seen have a rounded morphology. At 48 hours, both rounded and spindle-shaped cells are seen. By 96 hours, almost all cells seen have spindle morphology. (Original magnification, x20.)
Immunofluorescent Characterization of Hepatic Fibroblasts
To demonstrate that the spindle-shaped cells isolated from rat liver were in fact fibroblasts, we labeled the cells with a variety of markers. Fibroblasts would be predicted to express vimentin, but von Willebrand factor (marker of endothelia), smooth muscle actin (marker of smooth muscle and myofibroblasts), and cytokeratins (marker of epithelia) would not. As seen in Figure 2, putative rat fibroblasts stained for vimentin antibodies but not for von Willebrand factor, smooth muscle actin, and cytokeratin antibodies. We performed positive controls for these antibodies using the same methods on rat liver sections (for von Willebrand factor and smooth muscle actin) and cultured rat myofibroblasts (for smooth muscle actin). The present experiments confirm that spindle-shaped cells isolated from rat liver, as described above, are in fact fibroblasts.
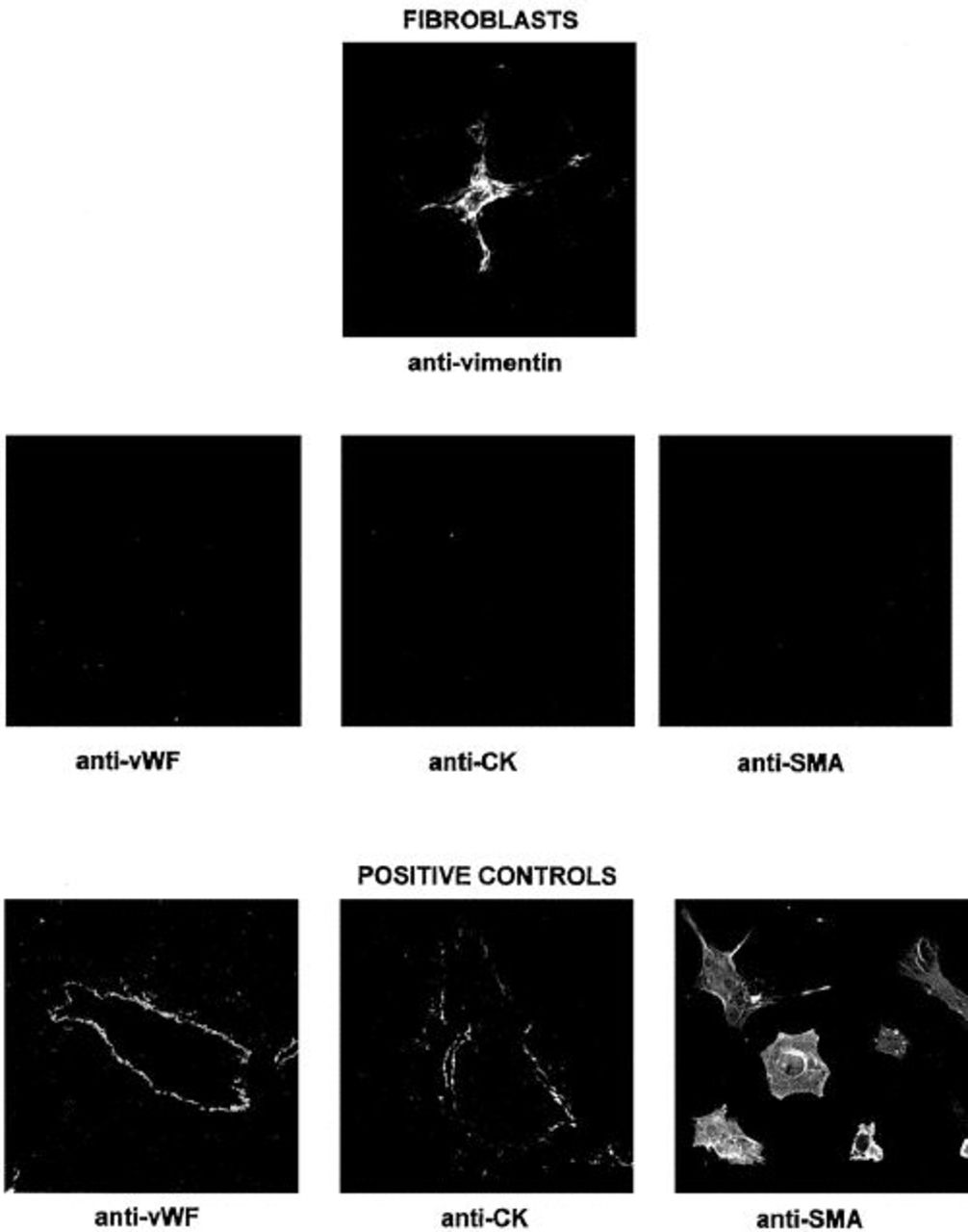
Putative rat fibroblasts do not stain with endothelial, epithelial, or smooth muscle markers. Confocal micrographs were obtained from putative rat fibroblasts, liver sections, and cultured rat myofibroblasts, as described in text. As is typical of fibroblasts, putative isolated rat fibroblasts stain with vimentin (original magnification, x120), but are negative (not shown) for von Willebrand factor (vWF; endothelial marker), cytokeratins (CK; epithelial marker), and smooth muscle actin (SMA; smooth muscle marker). Positive controls for von Willebrand factor are shown from a rat liver section labeling a central vein, and for CK from a rat liver section labeling an intrahepatic bile duct (original magnification, both x20). A positive control for SMA is shown from cultured rat myofibroblasts (original magnification, x120).
To demonstrate that these fibroblasts are actually a process of the isolation procedure rather than an artifact of the cell culture conditions, we observed the rat hepatic hilum preserved after collagenase digestion of hepatocytes that is used as the starting material for fibroblast isolation (Figure 3). The hematoxylin and eosin-stained section shows evidence of fibroblasts, epithelia, and endothelia. The hilum was stained for the markers noted above. Distinct populations of vimentin-, cytokeratin-, and von Willebrand factor-positive cells were identified, but no smooth muscle actin-positive cells were seen. These findings demonstrate that the cells identified were actually a process of the isolation procedure, in which they were selected by the culture conditions, because they were present in the starting material from which the cells were isolated.
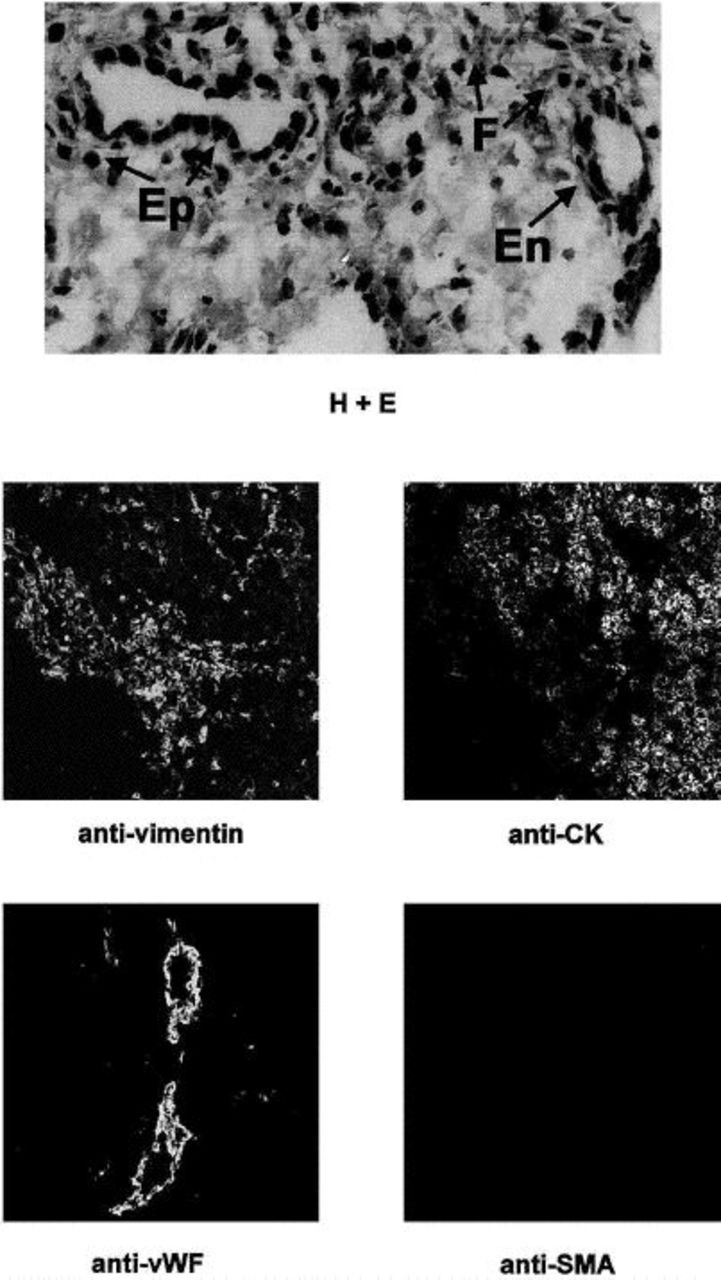
Rat hepatic hilum contains fibroblasts, epithelia, and endothelia, but not myofibroblasts. The top image is the hematoxylin and eosin-stained section of the rat hepatic hilum that was used as the source material for isolation of primary rat liver fibroblasts. Multiple cell types are seen, including fibroblasts (F), epithelia (Ep), and endothelia (En). The middle and bottom rows are confocal micrographs of rat hepatic hilum (original magnification, x20). Staining and imaging with markers detailed in the Figure 2 legend were performed using identical protocols. A section was double-labeled with antivimentin and anti-CK. As can be seen, vimentin-positive cells are distinct from those that expressed CK, which demonstrates that these cells are distinct populations. The bottom row shows endothelia within hepatic vessels that stained for von Willebrand factor. No sections stained positive for α-smooth muscle actin. Taken together, these data demonstrate that the rat hepatic hilum that is used as the starting material for fibroblast isolation contains distinct populations of fibroblasts, epithelia, and endothelia.
Electron Microscopic Characterization of Hepatic Fibroblasts
To further confirm that primary rat fibroblasts are indeed fibroblasts, we performed transmission electron microscopy on 96-hour cells. As seen in Figure 4, rat fibroblasts show no evidence of nuclear polarization, junctional complexes, microvilli characteristic of epithelia, pinocytic vacuoles, Weibal-Palade bodies, basal lamina, or filaments. These findings further support that putative primary rat liver fibroblasts are in fact fibroblasts.
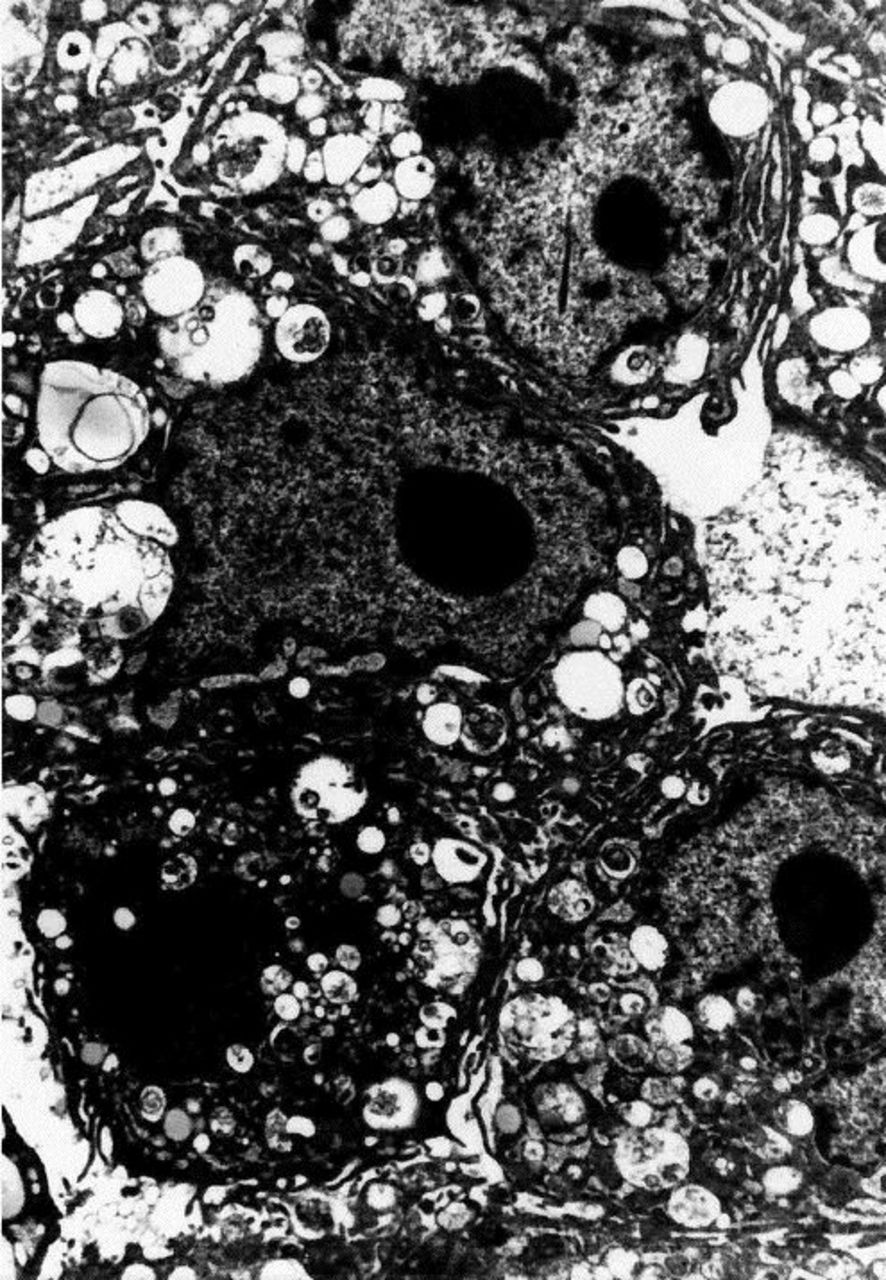
Photomicrograph showing primary rat fibroblasts that display typical findings under electron microscopy. We obtained transmission electron photomicrographs of primary rat fibroblasts using methods described in text. Three cells are seen in close proximity to one another. No evidence of nuclear polarization, basal lamina, junctional complexes, microvilli, Weibal-Palade bodies, pinocytic vacuoles, or myofilaments are seen, which demonstrates that primary rat fibroblasts are not of epithelial, endothelial, or smooth muscle lineage (original magnification, x3300),
Demonstration that Primary Hepatic Fibroblasts Express Procollagen
To test whether the cells isolated with the above-described methods were capable of synthesis, we performed RT-PCR for the presence of procollagen-1 (Figure 5). With oligonucleotides that are specific for procollagen-1, PCR products of the predicted size were present in cDNA from fibroblasts and myofibroblasts (positive control) but not in bile duct epithelial cDNA (negative control). These findings demonstrate that putative primary rat liver fibroblasts are in fact functional fibroblasts.
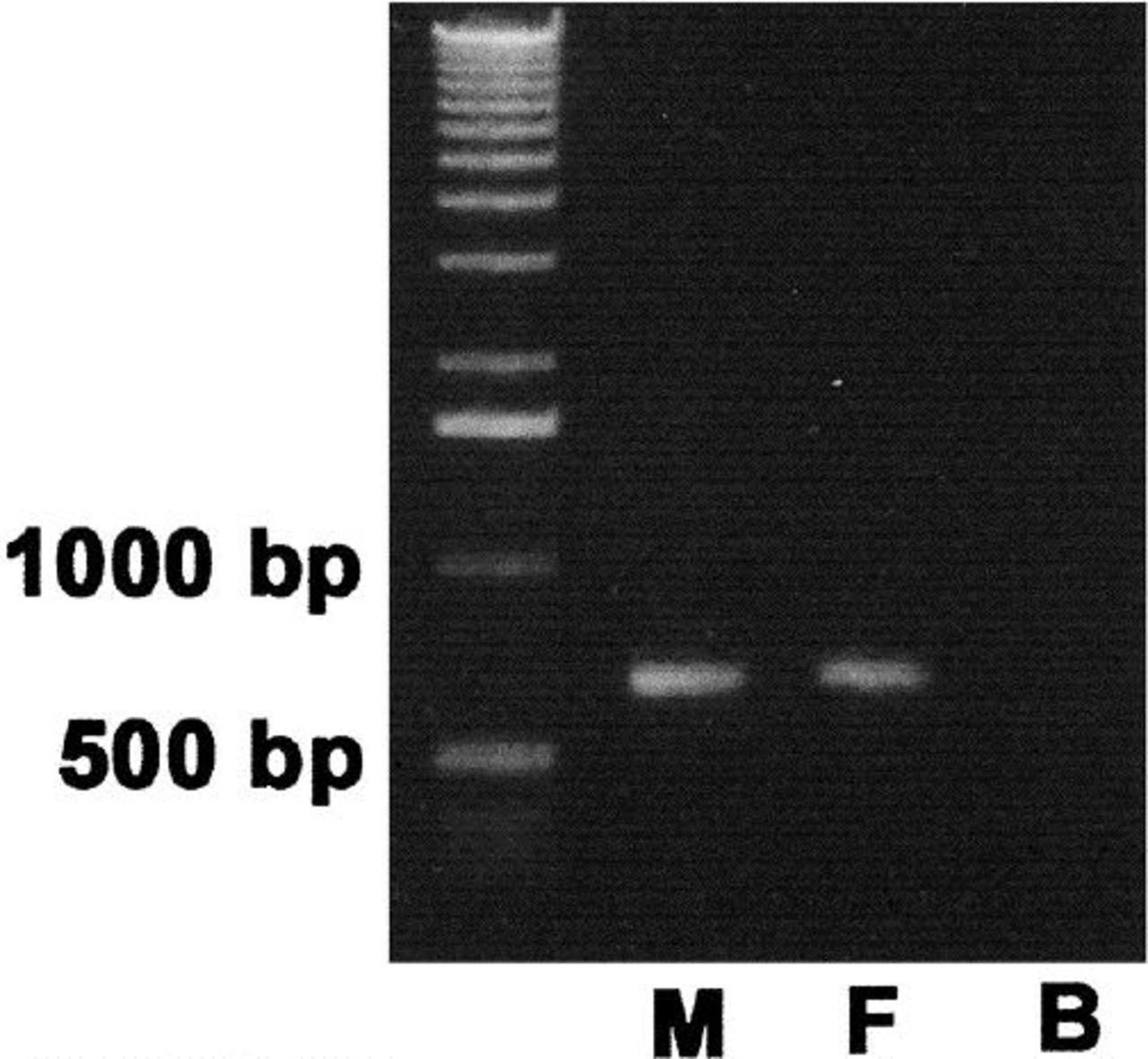
RT-PCR showing primary rat liver fibroblasts that express procollagen-1 mRNA. To determine whether rat liver fibroblasts were capable of functional collagen synthesis, we performed RT-PCR using rat procollagen-1-specific oligonucleotide primers. Bands of approximately 650 base pairs were observed from the positive control myofibroblasts (M) and from fibroblasts (F); however, no band was observed from the negative control bile duct epithelia (B).
Discussion
This article describes a method for the isolation of primary rat fibroblasts that is based on the method for isolation of nonparenchymal cells, which also has been used to isolate cholangiocytes and hepatic cells. We demonstrate that the cells that we have isolated are in fact fibroblasts because of their immunohistochemical staining profile, ultrastructural features, and functional expression of procollagen. The cells can be grown either in multiple-well plates for physiologic studies or on slides and/or coverslips for morphologic studies.
The isolation of various liver cell populations, including hepatocytes, 5 bile duct epithelia,2,3 and stellate cells, 4 has been one of the major advances in the understanding of liver physiology and pathophysiology of the past 20 years. For instance, isolation of hepatocytes and cholangiocytes has dramatically improved our comprehension of transport processes within the liver, 6 and isolation of stellate cells has led to a better understanding of hepatic fibrogenesis. 7 Although it has not been shown directly, it is clear from studies in other tissues, such as lung, that fibroblasts are important regulators of processes such as epithelial growth and differentiation. It is likely that fibroblasts in the liver also hold key signaling roles.
Is it necessary to isolate primary liver fibroblasts when both passaged and primary fibroblasts are available from a variety of tissues, such as skin? Although fibroblasts from cell sources other than the liver are useful experimental models, it is clear that fibroblasts are a heterogeneous population of cells both in physiologic8–10 and pathophysiologic states.11,12 One may surmise that while liver fibroblasts are indeed fibroblasts, there are significant differences between them and other models of fibroblasts that are used experimentally. In fact, fibroblasts are not defined morphologically but rather are defined as cells that synthesize collagen and extracellular matrix and that are not myofibroblasts or cells of other lineage. 13 Fibroblasts from various tissues may secrete different collagens and matrix components and may express a variety of heterogeneous proteins. Thus, only study of primary liver fibroblasts may be appropriate for studies of liver physiology.
The ability to isolate primary rat liver fibroblasts allows direct evaluation of the role of these cells in liver processes. Processes such as fibrogenesis and growth can be evaluated in in vitro models, and the role of fibroblasts as mediators of signaling may be understood better. Moreover, the role of fibroblasts in cell-cell interactions can be approached through co-culture experiments. In addition, fibroblast changes in pathological conditions will be elucidated through the isolation of fibroblasts in disease models.
Footnotes
Acknowledgments
The authors thank Lillemore Wallmark for technical assistance with confocal microscopy experiments, James Boyer for suggestions on cell isolation, and Rebecca Wells for her kind donation of cultured rat myofibroblasts.