
Abstract
Background:
Pityriasis rotunda is a rare cutaneous disorder characterized by scaly, circular, well-demarcated, hypo- or hyperpigmented, fine plaques over the trunk and extremities.
Objective:
We present a case of pityriasis rotunda in a 44-year-old African-Canadian woman who presented to a community dermatology practice in Toronto.
Results:
Pityriasis rotunda has been well described in Japan, Italy, and South Africa. It is extremely rare in North America, with nine reported cases to date, the majority of which were diagnosed in the United States.
Conclusion:
Pityriasis rotunda is a rare cutaneous disorder associated with systemic disease. To the best of our knowledge, this is the second report of pityriasis rotunda diagnosed in Canada.
Case Report
A 44-year-old Afro-Caribbean woman, who had emigrated to Canada from Jamaica, presented for assessment to a community dermatology practice in Toronto. She reported long-standing pruritic lesions involving her torso and extremities. She was otherwise systemically well. On examination, there were large hyperpigmented, scaly, circular, fine plaques over the abdomen, back, arms, and feet. The largest lesion was on the right hip (Figure 1), extending on to the abdomen. There was a subtle but sharp demarcation between the involved area and the normal surrounding skin (Figure 2).
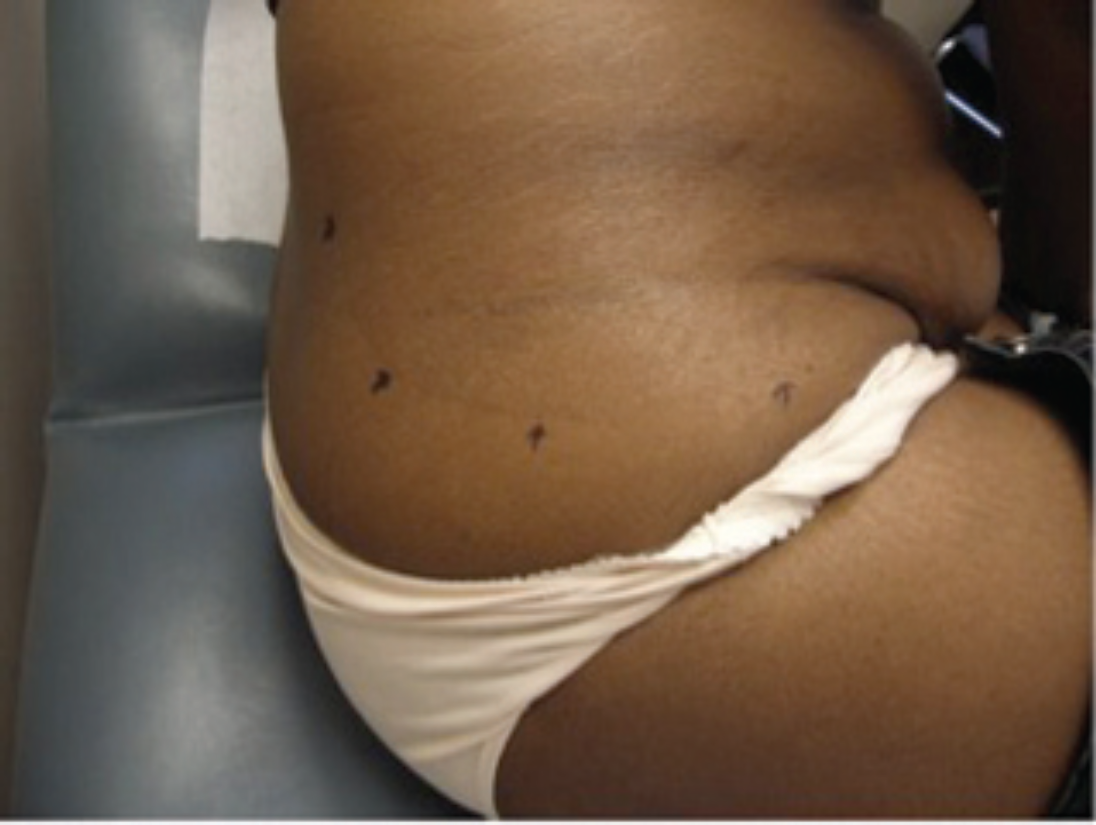
A 44-year-old African-Canadian with multiple hyperpigmented, scaly, thin plaques. The largest lesion, represented above, is overlying the hip.
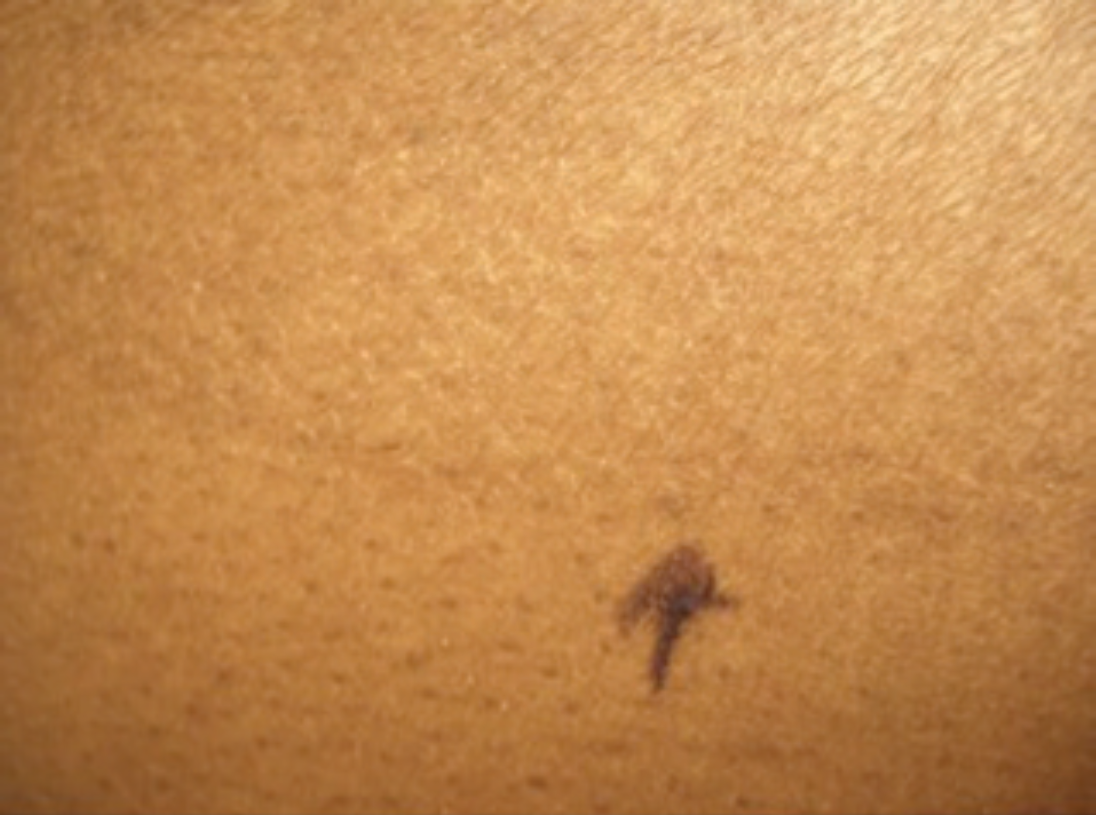
A sharp demarcation between the hyperpigmented scaly plaque involving the skin and the surrounding normal areas can be appreciated.
Based on the morphologic features, a provisional diagnosis of PR was made. A differential diagnosis of tinea corporis was considered. Skin biopsy revealed hyperpigmentation of the basal layer, a diminished granular layer, and hyperkeratosis consistent with PR (Figure 3). Periodic acid–Schiff staining was negative for hyphae. No fungi were detected by direct examination of a potassium hydroxide preparation or by culture.
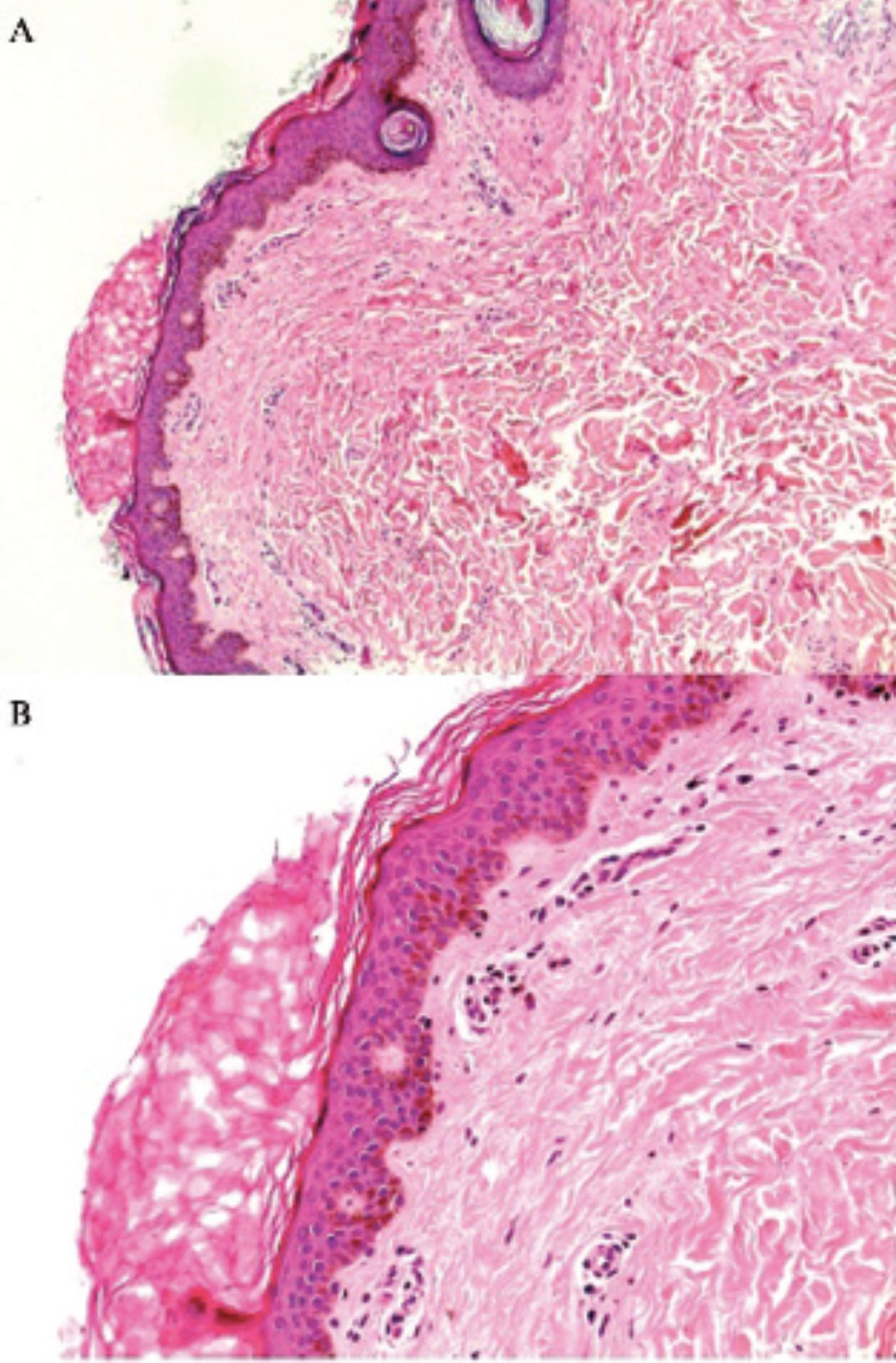
Hematoxylin-eosin staining showing hyperkeratosis without parakeratosis, a diminished granular cell layer, follicular plugging, increased pigmentation of the basal layer, and a mild perivascular lymphohistiocytic infiltrate (A, ×25 original magnification; B, ×100 original magnification).
A complete blood count, alkaline phosphatase, and aspartate transaminase were normal. Her α-fetoprotein was 9 μg/L (normal < 10 μg/L). Investigation for the possibility of underlying hepatic carcinoma was discussed with the patient, at which time, she mentioned that a test had been done on her liver previously. Discussion with her family physician revealed that an abdominal sonogram had been done the year before because of vague abdominal complaints, indicating an oval lesion, 5.5 × 4.2 cm in the right lobe of the liver, with a central echo-poor region, suggestive of a hemangioma. A computed tomographic scan confirmed the presence of the lesion, in keeping with a giant hemangioma with central necrosis. Subsequent follow-up by ultrasonography 5 months later showed the lesion to be stable in size. A Hepatology consultation together with further consultation with Radiology was arranged to be certain that a hepatic cancer was not being overlooked, in view of the presence of class I PR in a black patient. It was concluded that the lesion was a benign hemangioma. The lesions resolved over 3 months with conservative management with a lactic acid–containing moisturizer.
Discussion
PR remains a morphologically based diagnosis. The histopathologic features of hyperkeratosis and basal layer hyperpigmentation are well described. The pathogenesis of PR is unknown. Hashimoto and colleagues suggested a possible link with acquired ichthyosis. 6 Their group found a decrease in filaggrin and loricrin expression in affected areas. They did not find a corresponding difference in staining between affected and unaffected areas of keratin 1 and 10, antitransglutaminase, and antiinvolucrin antibodies. 6 Larger studies will be required to confirm this association.
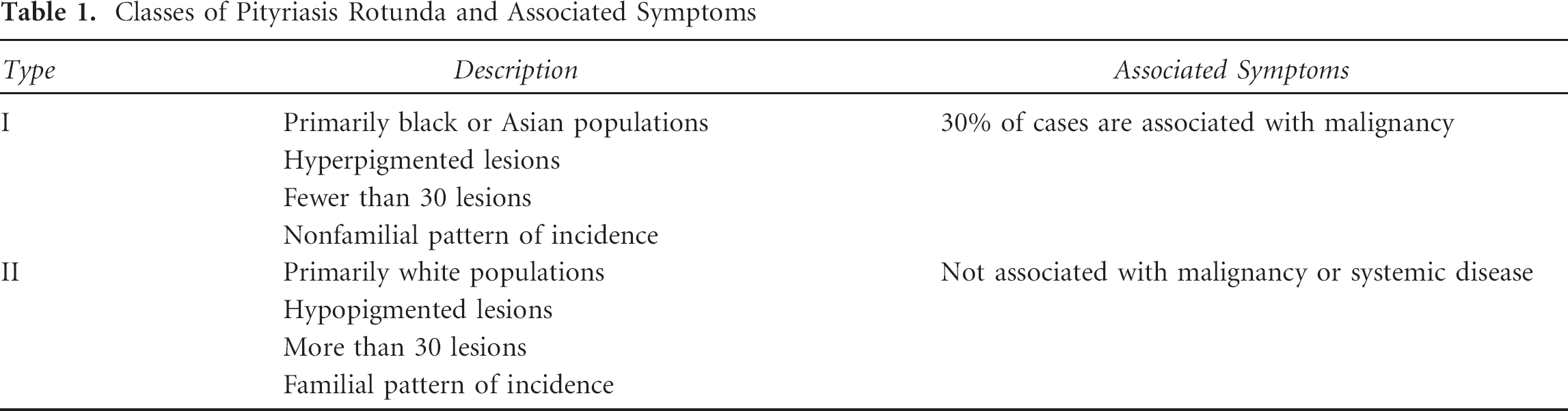
Grimalt and colleagues proposed a classification system for PR (Table 1). 7 Class I disease is defined as PR seen in black or Asian patients with less than 30 lesions. The plaques are hyperpigmented. This class is associated with systemic disease and malignancy, most notably hepatocellular carcinoma. Class II is defined as PR in white patients presenting with more than 30 lesions, usually hypopigmented. Class II is familial, and there is no association with underlying disease. 7
Classes of Pityriasis Rotunda and Associated Symptoms
Management of PR remains challenging. Although the literature indicates that use of topical retinoids, salicylic acid, and lactic acid ointments has been attempted, there is no known effective local therapy. 7 It is appropriate, however, to screen patients with type I PR for systemic disease. Treatment of the underlying disease has been associated with resolution of PR. 8 In type II PR, resolution may often be seen by adulthood; therefore, reassurance may be all that is required to manage patients and their families in those cases. 2
PR is well described in Japan, Italy, and South Africa. The initial case of PR was reported in the literature from Japan in 1906. 9 Since that time, there have been over 175 additional Japanese cases. 1 In Italy, PR has specifically been localized to the island of Sardinia, where 42 familial cases have been reported. 2 PR is also seen in the South African Bantu population, where particular associations with hepatocellular carcinoma and malnutrition have been reported. 3,–5
PR is very rare in North America. There have been six reported cases in the United States, all occurring in patients with black skin, including two American-born males. 10,,–13 To the best of our knowledge, this is the second report of PR to be diagnosed in Canada. 14
Footnotes
Acknowledgment
Financial disclosure of authors and reviewers: None reported.