
Abstract
Nephrogenic systemic fibrosis (NSF) is a rare condition that always occurs after acute or chronic renal failure with or without dialysis. The vast majority of cases in the literature are adults, and postmortem findings have been reported in only 5 cases. We report a 15-year-old male who developed NSF with multiorgan involvement after successful treatment of renal lymphoma and a subsequent sacral Ewing's sarcoma, and end-stage renal disease treated with hemodialysis. At autopsy, he was found to have diffuse dural osseous metaplasia, transmural bronchiolar fibrosis, diaphragmatic central tendon fibrosis, and fibrous plaques of the mitral valve. These previously unreported findings expand the spectrum of multiorgan involvement in NSF providing additional evidence that it is an emerging systemic disorder.
Keywords
INTRODUCTION
Initially described in 2000 [1], nephrogenic systemic fibrosis (NSF), previously known as nephrogenic fibrosing dermopathy, is a rare condition that occurs after acute or chronic renal failure with or without dialysis [2,3]. Progressive thickening of the skin usually begins in the lower extremities and may expand to involve the upper extremities and trunk. Joint contractures and progressive immobility are common [4]. Since the original description, cases with multiorgan involvement have been reported [5]. More than 170 cases have been reported [5,6]; however, postmortem findings have been described in only 5 cases [5,7–10]. Dural involvement has been identified in only 1 of these cases [8], and cardiac valvular disease and bronchiolar fibrosis have not been described. We report the postmortem findings in a 6th case. A teenage male requiring hemodialysis for chronic renal failure after successful chemotherapy and radiotherapy of sequential renal lymphoma and sacral Ewing's sarcoma developed NSF and progressive joint contractures subsequently complicated by multiorgan disease including dural osseous metaplasia, fibrous plaques of the mitral valve, and transmural bronchiolar fibrosis.
CASE REPORT
A monozygotic male twin presented at 3 years of age in renal failure and was diagnosed with bilateral renal diffuse large-cell lymphoma. Immunohistochemical studies revealed the tumor cells stained strongly positive for Pan-B markers. He was treated with hemodialysis and low-dose radiation therapy to his kidneys followed by chemotherapy including Adriamycin, methotrexate, vincristine, and 6-mercaptopurine. Renal function improved, and durable remission was achieved. Although he was able to stop dialysis, chronic kidney disease persisted, and he was treated with ferrous sulfate and epogen for anemia, calcium carbonate and calcitriol for renal osteo-dystrophy, sodium bicarbonate, and an appropriate diet.
At 11 years of age, he developed severe left leg pain and flexion contracture of his foot. Imaging revealed a mass at L4-S1 involving the left side of the sacrum, left sacroiliac joint, and left ilium; the spinal cord and its dura were not involved. Percutaneous biopsy revealed Ewing's sarcoma. The tumor stained positively for CD99 and negatively for CD3, CD20, and CD45RO. His estimated glomerular filtration rate was 14 ml/ minute/1.73 m2, and it was elected to start him on hemodialysis concomitantly with chemotherapy. The chemotherapy included vincristine, actinomycin D, cyclophosphamide, ifosfamide, and etoposide and lasted 10 months.
Bilateral ankle equinus contractures were identified after 8 months of chemotherapy for Ewing's sarcoma and 5 months after his 1st magnetic resonance imaging (MRI) scan with gadolinium (Gd). Previous computed tomography (CT) scans showed no evidence of fibrosis in these regions. At this time, his skin examination was unremarkable, and clinical, laboratory, and imaging studies provided no evidence of fibrosis other than the skin. He underwent bilateral Achilles tendon lengthening.
At 12 years of age, he developed severe secondary hyperparathyroidism because of non-compliance with dietary restrictions and phosphate binders. He was treated with total parathyroidectomy and forearm reimplantation of one of the glands. He developed hypocalcemia postoperatively from “hungry bone syndrome” [11] that resolved after 2 months of treatment with calcium supplements and vitamin D.
Also at 12 years of age, he developed a new onset seizure disorder that was treated initially with Neurontin and subsequently with Depakote and amitryptiline. At 13 years of age, he developed bilateral severe foot pain (considered then possibly secondary to the vincristine therapy) that subsequently progressed into foot contractures.
By 13 1/2 years of age, he had developed severe headaches that required administration of narcotics for pain control. An MRI scan revealed persistent meningeal enhancement, and a CT scan showed dural calcification. The serum calcium was normal.
His mobility and function deteriorated, necessitating multiple orthopedic procedures, and he became confined to a wheelchair. He started experiencing sustained hypercalcemia despite therapy with low-dialysate calcium, pamidronate, calcitonin, and steroids. The parathyroid remnant that had been implanted in his forearm was removed with no improvement of hypercalcemia. A head CT scan showed marked progression of dural calcification, as well as optic nerve sheath and carotid artery calcification (Fig. 1).
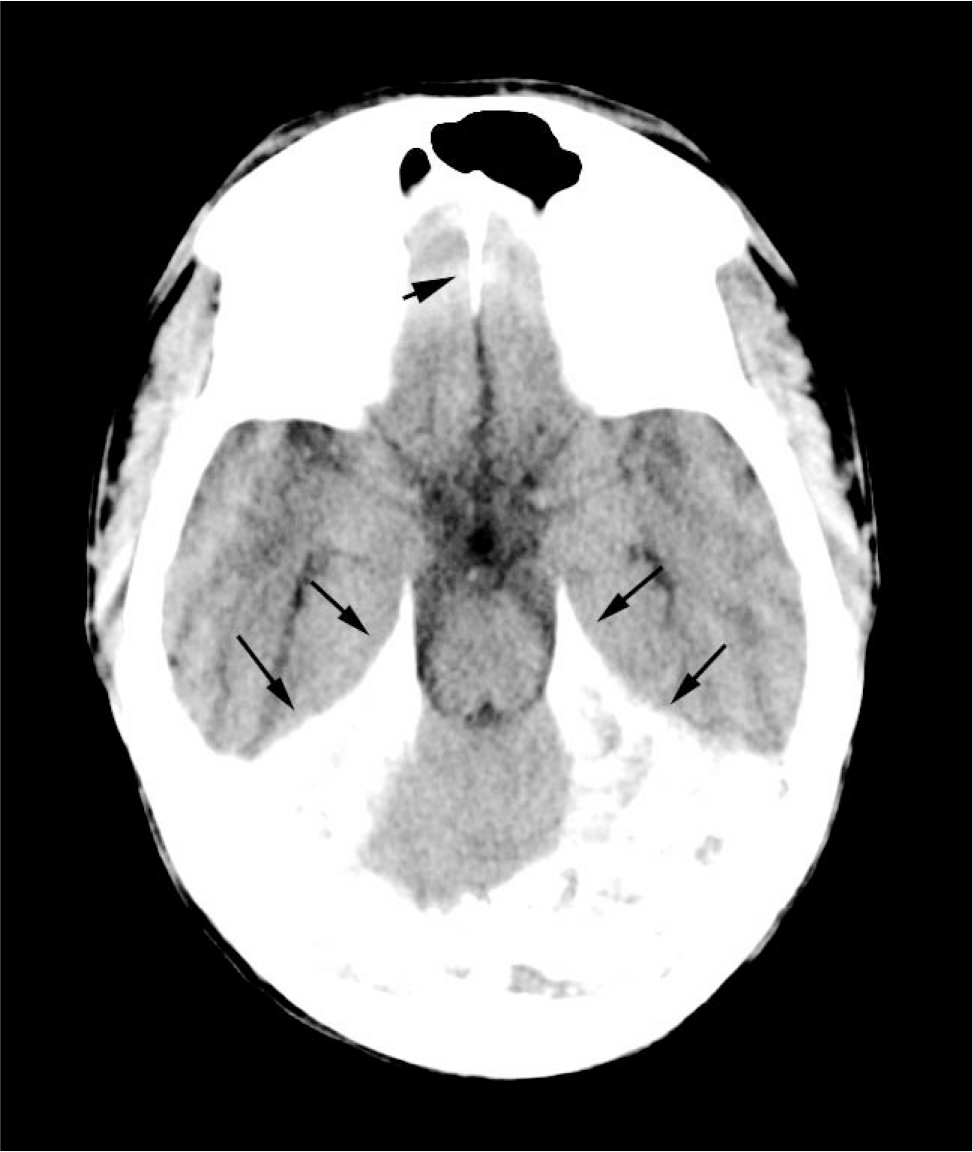
Axial computed tomogram of through tentorium demonstrates dense calcification (long arrows) and calcification in the falx cerebri (short arrow).
He then developed subcutaneous nodules, generalized xerosis, and a tight, sclerotic dermis of the distal extremities prompting a skin biopsy that confirmed the diagnosis of nephrogenic systemic fibrosis.
At 15 years of age, he was hospitalized to manage persistent limb pain, sacral ulceration, headaches, and distal extremity contractures. His localized pain progressed to total body discomfort. A gastrostomy tube was place for feeding. Five months before his death, funduscopic examination revealed sharp discs and no evidence of papilledema or hemorrhage.
His physicians felt that the NSF might improve with renal transplantation, and a transplant workup was performed. His coagulation screening revealed that he was heterozygous for the Factor V Leiden mutation and homozygous for the methyl-enetetrahydrofolate reductase C677T variant. Three months later, he received a living-related donor kidney transplant from his mother, but during surgery he experienced a junctional cardiac rhythm with bradycardia, hypotension, and cardiac arrest. This was not explained by electrolytes or calcium anomalies but might have been related to the high-dose methylprednisolone infusion given for immunosuppression [12–14].
He was resuscitated, but the newly transplanted kidney had minimal urine output. He was started on continuous venovenous hemodialfiltration, and heparin anticoagulation was used to try to prevent graft thrombosis. On the 2nd day after transplantation, however, a technetium-99m-mercaptoacetyltriglycine renal scan showed no perfusion to the transplant. After consultation with his family, the decision was made to withdraw further treatment. The dialysis was stopped after 4 days, and he was discharged to hospice and died.
Postmortem examination
The body was cachectic and revealed evidence of numerous surgical procedures aimed at correcting severe joint contractures of his extremities. The muscles of the extremities were markedly atrophic. Residual neoplasia was not identified grossly or microscopically. The skin, especially over the lower legs and feet, was pigmented, thickened, and focally excoriated. The skin biopsies taken 5 1/2 months before death and at the autopsy were essentially identical, revealing severe proliferation of spindled cells throughout the papillary and reticular dermal fibrosis with extension into the subcutaneous septa (Fig. 2). Elastic fibrils were focally increased and thickened. Mucin was not increased within the dermis. The epidermis was severely atrophic. Inflammatory infiltrates, necrobiosis, and mucin accumulation were not present. The exceedingly thickened and calcified dura encased the brain; the tentorium was a rigid plane of tissue (Fig. 3). Microscopic examination revealed severe fibrosis, as well as extensive osseous metaplasia and dystrophic calcification (Fig. 4).
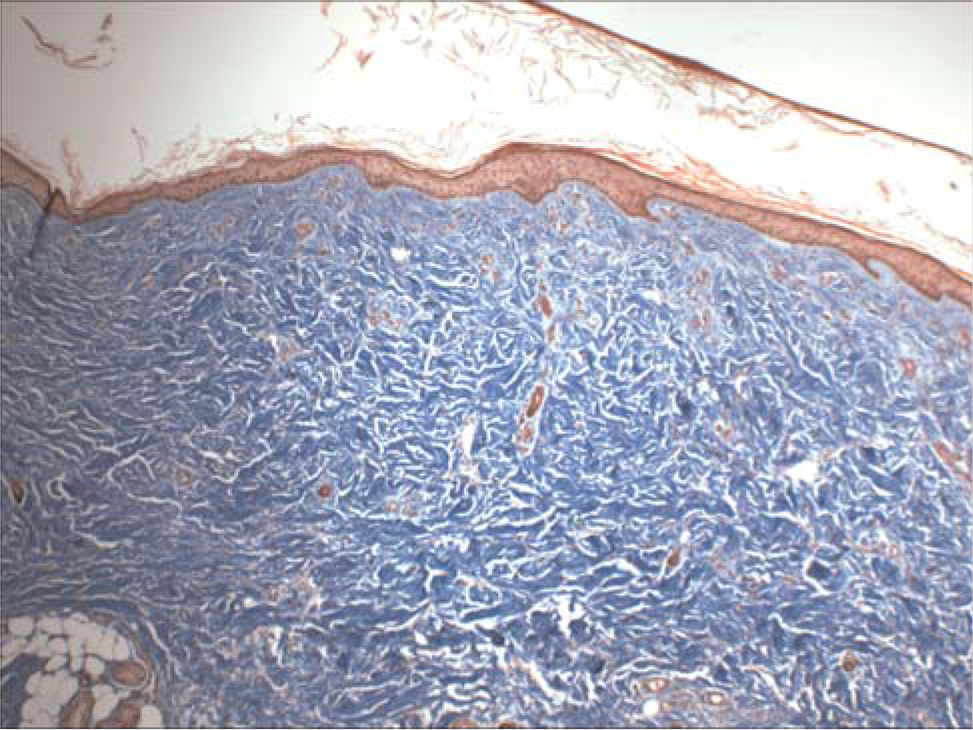
Postmortem skin demonstrates severe fibrosis of the reticular and papillary dermis in the absence of inflammation or deposits of mucin. The epidermis is severely atrophic. H&E ×100. A color version of this figure is available in the online journal.
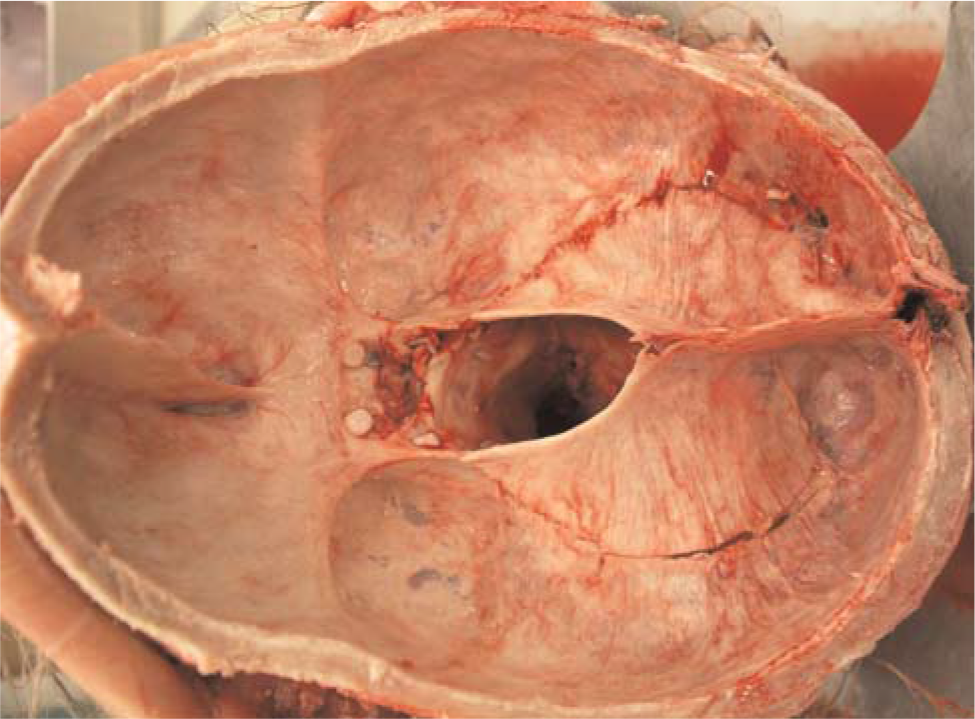
The dura is severely thickened; the tentorium is a rigid plane composed of dense fibrous tissue and osseous metaplasia. A color version of this figure is available in the online journal.
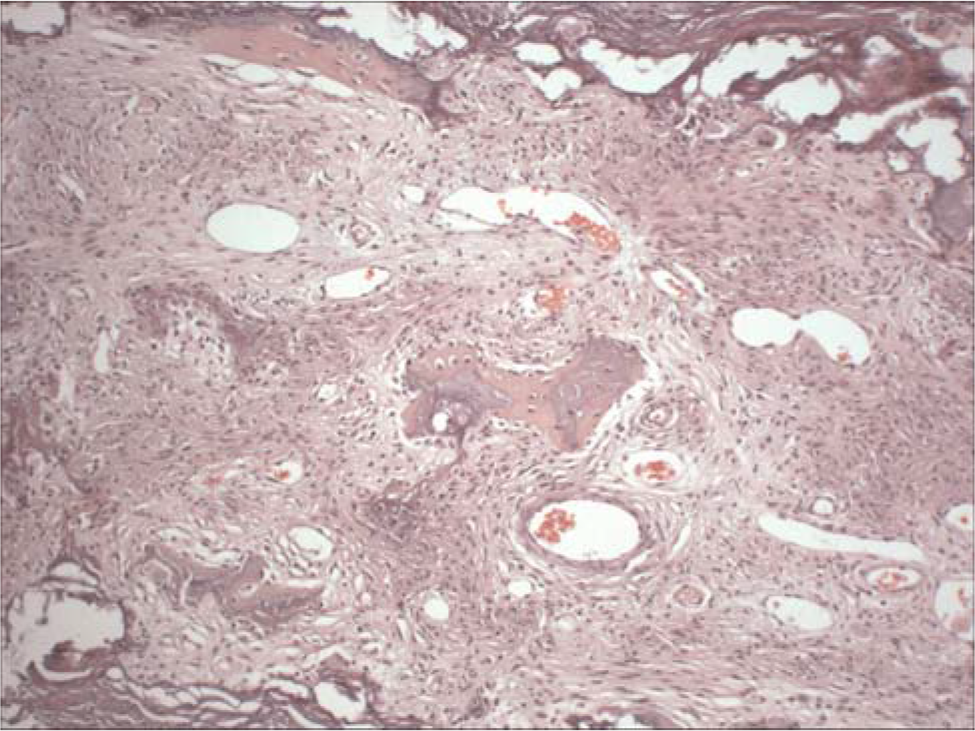
The tentorium demonstrates severe fibrosis and osseous metaplasia. Dystrophic calcification is also present. H&E ×100. A color version of this figure is available in the online journal.
The lungs were severely congested and displayed diffuse moderate pleural and interlobular septal fibrosis; the bronchioles revealed moderately severe transmural fibrosis (Fig. 5). Calcium and iron were deposited focally along elastic membranes of proximal arteries and bronchi. The central tendons of the diaphragm displayed bilateral severe fibrosis and rigidity (Fig. 6). Microscopically, dense fibrous tissue extended from the central tendons into the adjacent skeletal muscle tissue.
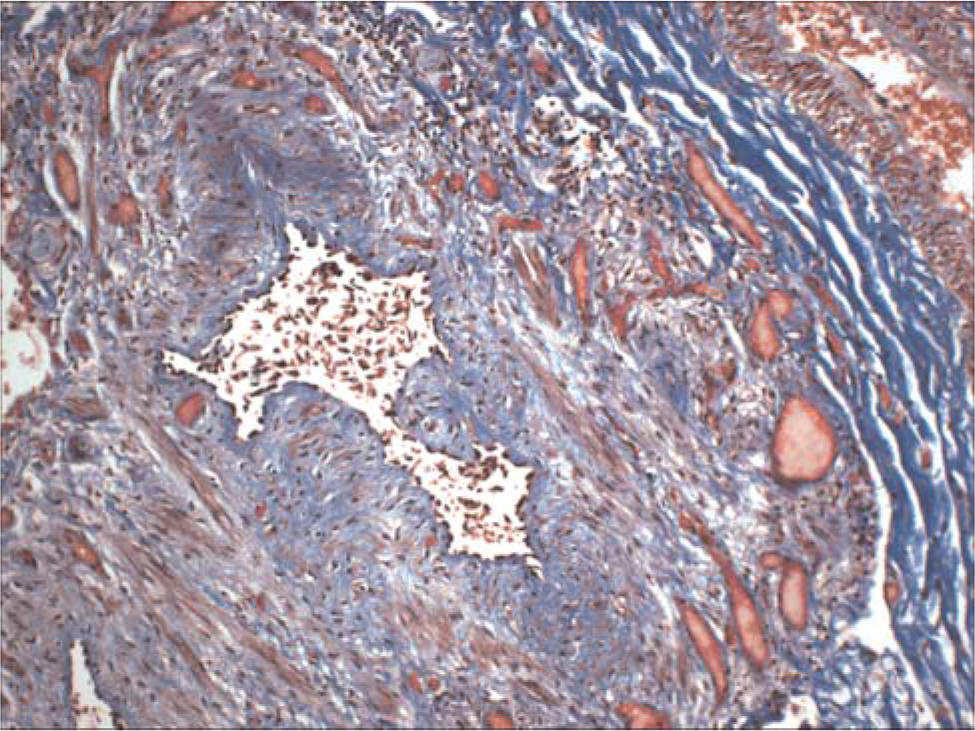
A bronchiole demonstrates severe transmural fibrosis. The accompanying pulmonary artery is at the upper right. Masson Trichrome ×100. A color version of this figure is available in the online journal.
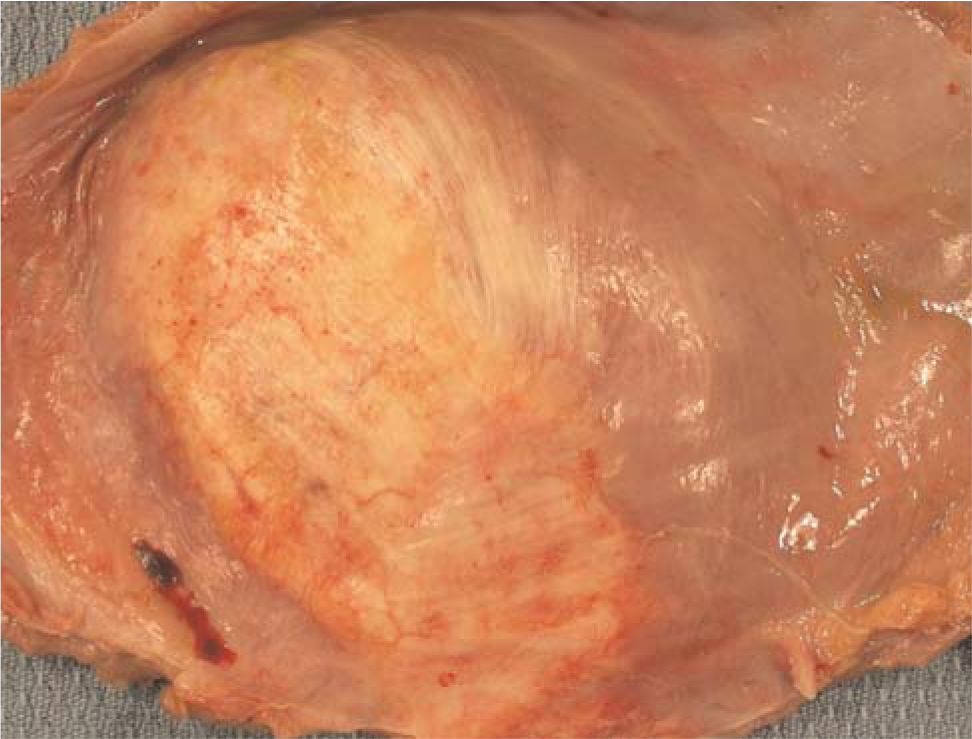
Severe fibrous thickening of the central tendon extends into the muscle of the diaphragm. A color version of this figure is available in the online journal.
The mildly enlarged heart revealed thick fibrous plaques of the anterior and posterior mitral leaflets (Fig. 7). The other cardiac valves, myocardium, and endocardium were only mildly fibrotic. An intravenous catheter extended through an occlusive organized thrombus that extended from the inferior jugular vein into the superior vena cava and proximal right atrium. Thromboembolization was not found in other organs.
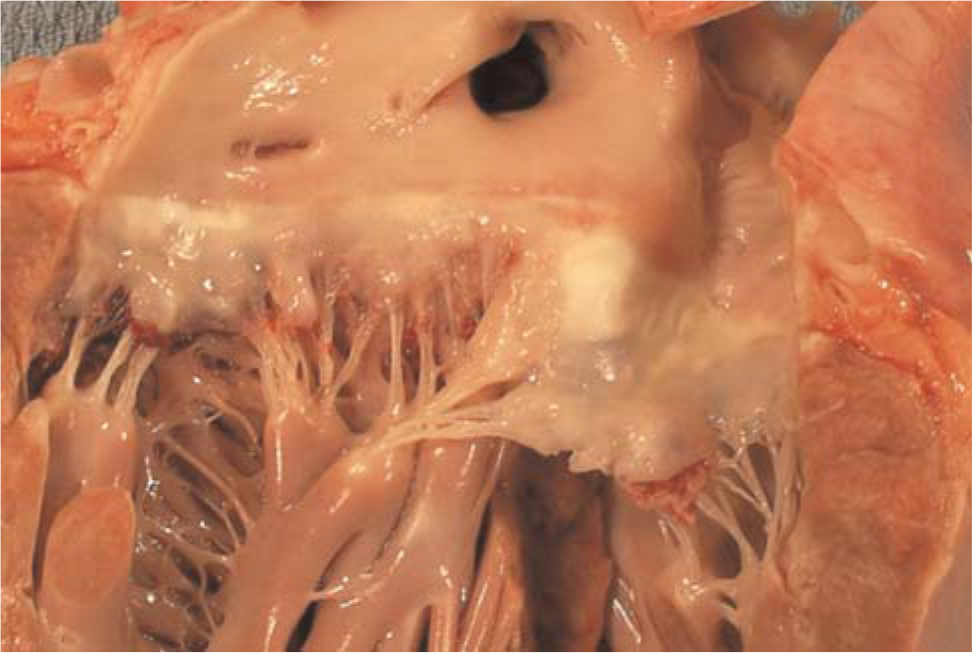
The anterior and posterior leaflets of the mitral valve are altered by thick fibrous plaques. The left ventricle displays mild hypertrophy reflected by rounding of the basal margin. A color version of this figure is available in the online journal.
Acute and chronic biventricular cardiac failure was evidenced by severe pulmonary congestion and numerous intra-alveolar sidero-phages, and hepatic panlobular congestion, bridging centrilobular degeneration, and necrosis.
One of the small, end-stage native kidneys revealed a circumscribed region of acquired cystic change [15]. The transplanted kidney revealed severe hemorrhagic congestion and necrosis; the vascular and urinary surgical anastamoses were intact, and the renal artery and vein were not occluded by a thrombus.
Immunochemistry findings
CD-34 and procollagen I results for the skin and extracutaneous sites are shown in Table 1. Immunohistochemical stains were generously per-formed by Dr Shawn Cowper, Yale Dermatopathology Laboratory.
Immunohistochemical results

DISCUSSION
Postmortem findings have been reported in only 5 cases of NSF [5,7–10]. We now add a 6th case, a teenage male with NSF that developed after successful chemotherapy and radiation for bilateral renal lymphoma followed by chronic renal failure ultimately necessitating chronic hemodialysis. Seven years later, he developed a left sacroiliac Ewing's sarcoma that was also successfully treated with chemotherapy and radiation. Our case had unique and previously unreported findings of diffuse dural fibrosis and osseous metaplasia, transmural bronchiolar fibrosis, and fibrous plaques of the diaphragmatic central fibrous tendon bilaterally and anterior and posterior mitral leaflets.
Our patient presented at 3 years of age with a bilateral diffuse large-cell lymphoma of B-cell type associated with renal failure. This is similar to a previous report describing a 6-year-old girl presenting with renal failure secondary to bilateral T-cell lymphoma [16]. Skin changes were not observed after chemotherapy and low-dose radiation to his kidneys. Despite persistence of renal failure, our case recovered sufficiently to discontinue hemodialysis for nearly 8 years until its reinstitution for end-stage renal disease. At 11 years of age, he developed a left sacroiliac Ewing's sarcoma near the field of prior radiation therapy for his renal lymphoma. Given the rarity with which Ewing's sarcoma has followed radiation therapy for a primary malignancy [17], we cannot exclude its significance in our case.
The death of our patient was caused by withdrawal of dialysis treatment decided by the patient and his family as a result of the extremely prolonged painful medical course, despite intensive maximum pain management therapies.
All cases of NSF have had a history of acute or chronic renal failure, and most had undergone dialysis. Most patients with NSF are adults; the mean age is nearly 50 years, and ages range from 8 to 81 years [18]. Approximately half of the cases are male. One had been awaiting a liver transplant after developing end-stage renal disease secondary to idiopathic membranous nephropathy [19], and another had systemic lupus erythematosus complicated by rapidly progressive glomerulonephritis [20]. In these reported cases, NSF presented clinically with progressive cutaneous fibrosis evolving into loss of mobility and joint contractures affecting initially the lower extremities with progression to the thighs, upper extremities, and finally the trunk. Facial involvement is rare; eye involvement has been reported but was not observed in our patient. Patients with NSF had received a variety of medications, including immunosuppressive drugs and erythropoietin [3,18].
Five pediatric cases have been reported. In contrast to our case, all 5 were living at the time they were reported. They include an 8-year-old male with type-II membranoproliferative glomerulonephritis, a 9-year-old male with focal segmental glomerulosclerosis, a 16-year-old female with branchio-otorenal syndrome, a 17-year-old male treated with peritoneal dialysis for failure of a renal transplant at 2 years of age [21], and a 19-year-old male with multicystic dysplastic kidney disease [22,23]. All developed renal failure for which they were treated with hemodialysis and renal transplants, NSF developed in all after onset of renal failure, and the joint contractures varied in severity.
Extracutaneous disease is being increasingly recognized in NSF. Disease has now been reported in the skeletal muscles, including the diaphragm and psoas, as well as in the pericardium, myocardium, great vessels, pleura, lungs, renal tubules, dura, rete testis, and tunica albuginea [5,7,8,10,18,24]. Our case expands the spectrum of extracutaneous disease associated with NSF by its involvement, severity, and/or presence of osseous metaplasia of the dura (Fig. 3), mitral valve (Fig. 7), bronchioles, and central tendons of the diaphragm (Fig. 6). Gibson and colleagues [8] reported “significant thickening” but did not describe the extreme rigidity and osseous metaplasia of the dura encasing the brain seen in our case. Bronchiolar transmural fibrosis observed in our case has not been reported previously; clinical evidence of restrictive or obstructive airway disease was not identified, and autopsy did not reveal alveolar distention or lipoid pneumonia. Fibrosis of the diaphragmatic muscle has been described, but fibrous plaques affecting primarily the central tendons were the dominant finding in our case; microscopic examination revealed severe fibrosis and a few multinucleate giant cells. The anterior and posterior mitral leaflets were altered by calcified fibrous plaques, but clinical valve dysfunction had not been identified.
The characteristic pathologic finding of the skin is severe fibrosis with abundant collagen. Osteoclast-like giant cells and dystrophic calcification have also been described, as seen in the diaphragm of our case [25]. Hauser and colleagues [26] described fibroblastic proliferation and increased collagen with extracellular mucin evolving several weeks later into pure sclerosis. Our case revealed severe fibrosis without inflammation or mucin in both the surgical and the postmortem skin biopsies. Sequential skin biopsies have been evaluated in only a few cases and have showed little change over time.
The diagnosis rests upon characteristic histologic findings in the skin in addition to labeling of the proliferating spindle cells in the dermis with CD34 and procollagen I as documented in our case. Zones of fibrosis in extracutaneous sites show similar histologic findings but have been only rarely subjected to immunohistological analysis. Daram and others [7] reported a 39-year-old male with end-stage renal disease caused by hypertension and treated with hemodialysis; at autopsy, spindled cells within the fibrotic regions of the pleura, diaphragm, pericardium, tunica, albuginea, and tissue overlying the pulmonary trunk were CD34 and CD45RO positive. Staining of these sites for procollagen I was not undertaken. The extracutaneous autopsy tissues in the case reported by Jimenez and colleagues [9] were not evaluated for CD34 or procollagen I. The bronchioles, diaphragm, myocardial interstitium, and endocardium stained intensely for both CD34 and procollagen I in our case; the dural fibrous tissue stained for procollagen I but not CD34. These immunohistochemical findings in our case suggest that pathogenesis of the extracutaneous fibrosis was similar to that in the dermis and subcutis.
The pathogenesis of NSF is not completely understood. The presence of increased fibroblasts, collagen and CD34, and procollagen I–positive spindled cells [7,26–28], CD45RO [7,28], CD68 [7,26,27], and TGF-β [9] indicate the importance of fibrogenic cytokines [3], derived from the bone marrow fibrocytes [26]. In cases with osseous metaplasia, such as ours, it has been suggested that altered matrix dysregulation due to altered TGF-β, metalloproteinases, and activation of osteoclasts may account for its pathogenesis [25].
Recent work suggests Gd exposure during MRI exams may induce NSF. Grobner reported that 5 of 9 cases with end-stage renal disease treated with dialysis developed NSF 2 to 4 weeks after undergoing magnetic resonance angiography using Gd-containing contrast agent [6,29]. Our patient underwent 20 MRI studies, 13 of them enhanced with Gd, which may have contributed to the severe progression of his disease. Gadolinium, the half life of which may be prolonged up to 130 hours in renal failure, can form precipitates with phosphate and carbonate that are deposited in skin and muscle, as well as other tissues causing initial mild inflammatory infiltration with TGF-β expression [30]. As a result, it has been recommended that “use of Gd be excluded to the greatest extent possible in patients with renal disease who are acidotic [31].” The formal recommendation of the Food and Drug Administration is present at http://www.fda.gov/cder/drug/advisory/gadolinium_agents.htm.
We could find no reports linking NSF with chemotherapy or occurring in the absence of chronic renal failure. However, there are reports of progressive systemic sclerosis (scleroderma) associated with Hodgkin's lymphoma treated with ABV and with cyclophosphamide, doxorubicin, vincristine, and prednisolone [32]; non-Hodgkin's lymphoma treated with cyclophosphamide, adriamycin, and prednisone [33]; and breast cancer treated with doxorubicin and cyclophosphamide [34]. Although it is impossible to prove that the skin disease and contractures in our case were not related to his chemotherapy, it seems very unlikely considering he had received a 1st course of chemotherapy 7 years before with no subsequent skin changes. The head, chest, and abdominal CT scans obtained at the time of diagnosis of the 2nd malignancy were normal with no evidence of fibrosis. The feet contractures were noted during the 2nd course of chemotherapy after the start of dialysis, which leads us to believe it was the 1st sign of NSF rather than an unusual side effect of the chemotherapy.
Osseous metaplasia has been reported in only 1 case, and it was in the skin [35]. In our case, this metaplastic change was present only in the dura. Interestingly, osseous metaplasia of the dura has been described in dogs after spinal cord degeneration (which was not present in our case), in the use of nonabsorbable dural patch material derived from bovine pericardium, and in association with a meningeal osteosarcoma in dogs as well as humans after spinal cord trauma [36–39].
A variety of laboratory abnormalities, including elevation in erythrocyte sedimentation rates and C-reactive protein, have been reported, but they are inconsistent [3].
In our case, an organizing thrombus extended from the superior vena cava into the proximal right atrium that we interpreted as a complication of the chronic placement of the dialysis catheter in the right jugular vein terminating in the right atrium; embolization to the lungs and other organs did not occur. Nevertheless, this raises the question of an underlying coagulopathy, especially given the identification of Factor V Leiden mutation. In this regard, Jain and colleagues [23] reported a 9-year-old male for whom the suspected cause of death was pulmonary embolism; however, an autopsy was not performed. Although Factor V Leiden mutations were not detected, low levels of protein C, protein S, and antithrombin III were identified and interpreted as a result of urinary protein loss. The 2nd case reported by Jain and others was a 19-year-old male with renal failure secondary to renal multicystic dysplasia; he also had a history of homocysteinemia and protein C deficiency resulting in a hypercoagulable state, yet his clinical course had not been complicated by vascular thromboses. The Factor V Leiden mutations were not identified in this patient either. The Factor V Leiden mutation found in our case may have played a role in the graft loss and in the thrombosis associated with the catheter; however, it must be emphasized that these complications can also occur without any underlying coagulation defect. The absence of thromboembolization in other organs suggests that the mutation was not clinically significant in our case.
Severe and rapidly progressive skin disease correlates with poor prognosis and death. Additionally, clinical evidence of multiorgan involvement appears to portend a much poorer prognosis compared with disease limited to the skin [5,8,9].
Several treatment modalities, including cyclophosphamide [20], interferon [40], immunoglobulins [19,41], UV-A1 phototherapy [42], photodynamic therapy [43], plasmapheresis [40,44,45], and extracorporeal photopheresis [46], have been attempted. The patient treated with cyclophosphamide showed no progression of his skin disease after 8 months [20]. Skin disease has improved in some treated with UV-A1 phototherapy [42] or extracorporeal photopheresis [47]. Improved joint mobility after photopheresis therapy has also been reported [46]. The 17-year-old male recently reported by DiCarlo and colleagues [21] experienced near complete resolution of active cutaneous lesions with topical therapy and compression stockings combined with 1 course of pulsed intravenous methylprednisolone and a short course of weekly methotrexate. In the final analysis, no treatment has been shown to be consistently effective [6]. In their review of the literature to which they added 12 cases of their own, Mendoza and colleagues [18] reported that only 20% of the patients showed modest improvement, whereas 28% did not improve and 28% died. It should be emphasized, however, that this review did not provide objective or reproducible measures of improvement, nor were causes of death listed for the 28% who died. Therefore, future morbidity and mortality studies should be aimed at comparing NSF cases to matched controls.
In conclusion, we agree with the view expressed by Cowper and Boyer [29] that NSF “is an emerging systemic fibrosing disorder” that always occurs in association with renal failure. As such, it represents an even greater therapeutic challenge than when limited to the skin. More information may be found at the Nephrogenic Fibrosing Dermopathy website [31].