
Abstract
An 8-month-old boy presented with a 6-week history of a skull mass of the anterior fontanelle. The mass was excised, and the histopathologic features were diagnostic for melanotic neuroectodermal tumor of infancy. The tumor showed focal myogenin positivity, which has not been previously reported in this tumor. The patient has no evidence of recurrent tumor 10 months after the excision. No adjuvant therapy was given. In addition, we stained a case of melanotic neuroectodermal tumor of infancy obtained from Columbus Children's Hospital; focal myogenin positivity was present.
INTRODUCTION
Melanotic neuroectodermal tumor of infancy (MNTI) is a rare neoplasm of neural crest origin that often occurs during the 1st year of life. The tumor is rapid growing and usually benign. It has a high recurrence rate, and a small number of cases are malignant [1]. The majority of cases involve the maxilla. Other sites include the skull and mandible. Unusual sites include the epididymis, mediastinum, scapula, ovary, uterus, and femur [2]. MNTI consists of a bimodal population of cells with primitive neural or melanotic differentiation. As such, the tumor cells predictably stain positively for neuron-specific enolase, synaptophysin, cytokeratin, and HMB45, with occasional staining for S-100, neurofilament, and chromogranin. Unexpected reactivity for muscle-specific actin, desmin, glial fibrillary acidic protein, CD57, and epithelial membrane antigen has also been reported in some cases, indicating that these are polyphenotypic lesions [2].
We report a case of MNTI in an 8-month-old boy who presented with a skull mass involving the anterior fontanelle. The tumor showed focal positivity with the myogenin stain. We stained an additional case of MNTI from a 3-month-old patient with a maxillary tumor that also showed focal myogenin positivity. Although markers of muscle differentiation (actin and desmin) have been previously reported in a few cases of MNTI, to our knowledge, this is the 1st report of myogenin positivity.
CASE REPORT
The patient had no skull abnormalities at birth. At approximately 6–1/2 months of age, he was noted by his mother to have a small bump at the anterior fontanelle that rapidly enlarged during the next 6 weeks. On physical examination, a 5 × 5-cm mass was noted; the skin was mobile over the lesion and appeared to be fixed to the skull. The physical examination was otherwise unremarkable. One month later, the skin became adherent to the mass and appeared erythematous at the center of the lesion. A skull radiograph showed a soft-tissue mass accompanied by adjacent calvarial sclerosis and thickening overlying the anterior fontanelle (Fig. 1). Magnetic resonance imaging revealed a vascular lesion extending through the calvarium and depressing a patent sagittal sinus; a vein was visualized that extended from the sinus to the mass. A craniotomy was performed with excision of the mass within a 2-cm margin. The mass involved the skin and cranium and invaded the dura. Dural reconstruction was performed, and the skin defect was closed by a plastic surgeon.
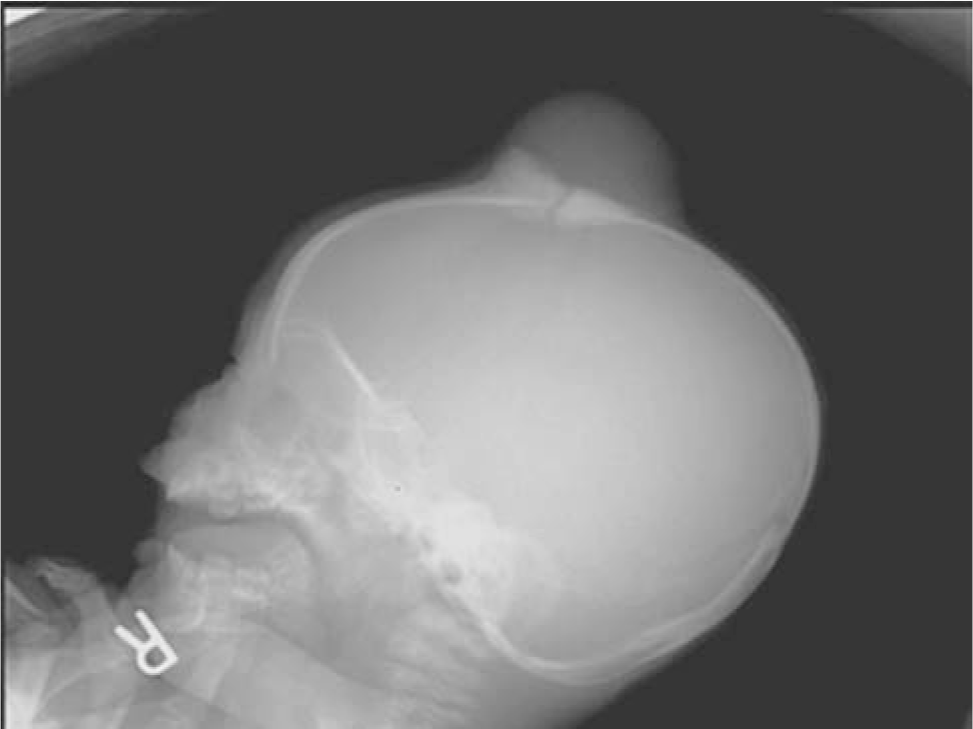
Skull x-ray shows a soft-tissue mass accompanied by adjacent calvarial sclerosis and thickening overlying the anterior fontanelle.
The tissue consisted of a saucer-shaped fragment of bone measuring 8.0 × 7.2 × 2.3 cm (Fig. 2). There was a central defect containing tumor, which measured 5 cm in diameter. An ellipse of skin and subcutaneous tissue measuring 5.5 × 4.8 × 2.5 cm was separately submitted. The tumor consisted of a firm grey mass with areas of brown discoloration (Fig. 3).
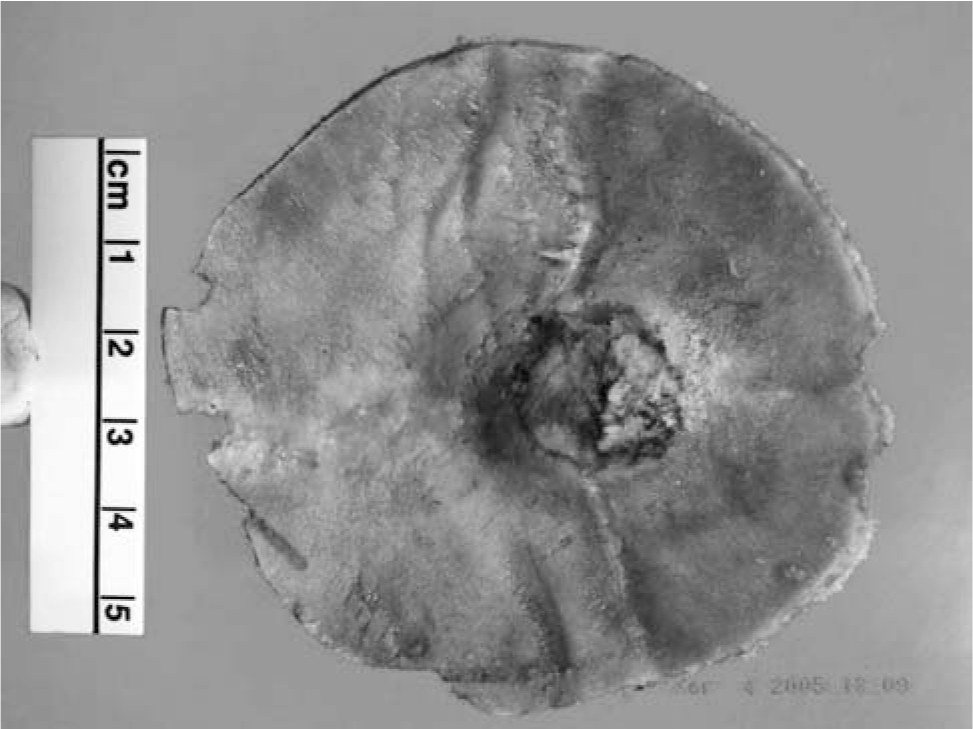
The skull from resection shows a central defect with tumor invasion.

The scalp from resection shows sclerosis and brown pigment.
Microscopically, the tumor was composed of nests of cells extending from the dermis of the scalp into the bone of the skull. The tumor cell nests contained small, discohesive cells with hyperchromatic nuclei and small amounts of eosinophilic cytoplasm. Mitoses were seen in these cells. At the periphery of the tumor cell nests were larger epithelioid cells, some of which contained brown pigment. The epithelioid cells had fine chromatin with prominent nucleoli. A dense fibrous stroma with bands of collagen was associated with the tumor cells (Fig. 4). Immunohistochemical stains showed that the epithelioid cells were positive for HMB45, neuron-specific enolase, and wide screen keratin. Some of the epithelioid cells were positive for S100. The small undifferentiated cells stained with neuron-specific enolase, with a few scattered cells positive for myogenin (Fig. 5). Desmin, CD99, and GFAP stains were negative. Cytogenetics revealed a normal karyotype.
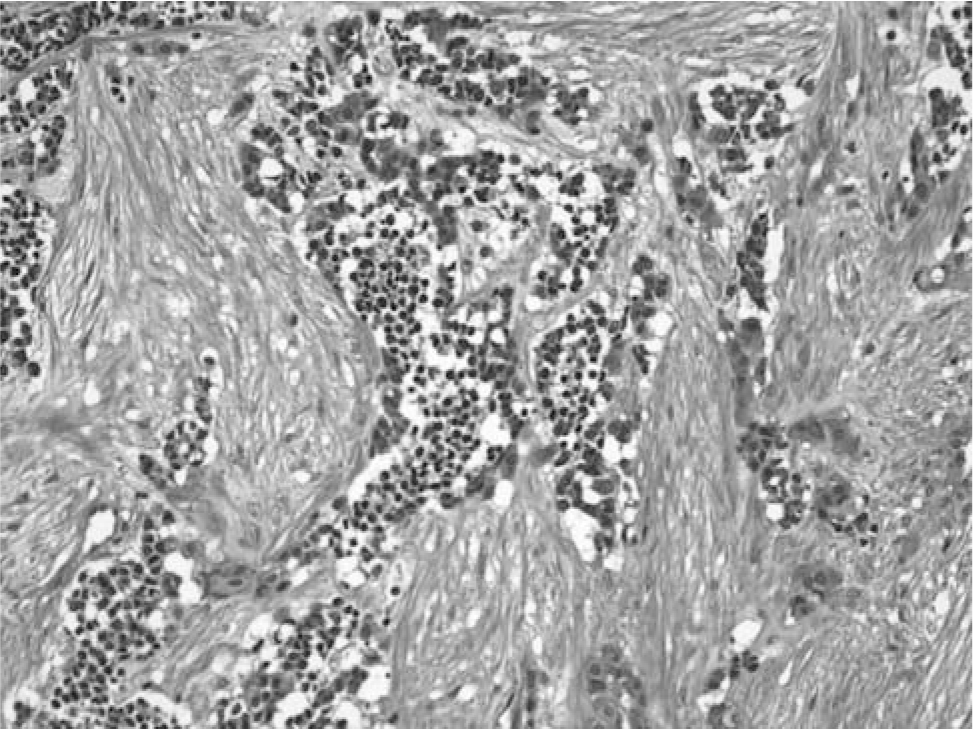
The hematoxylin and eosin stain shows a nest of pigmented epithelial cells surrounding neuroblasts in a dense stroma.
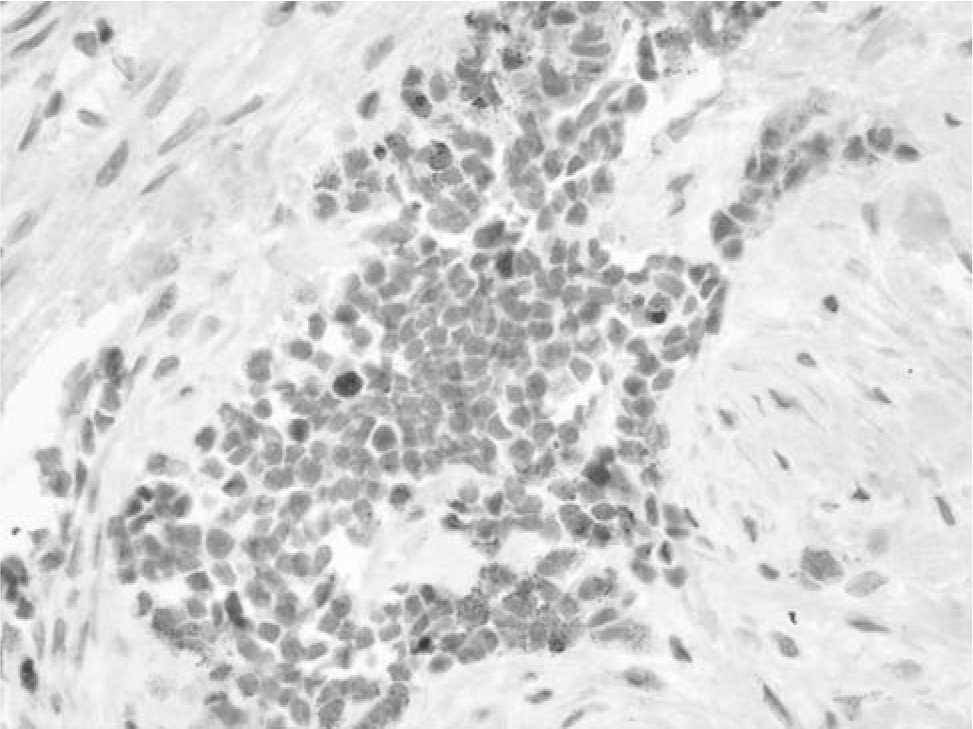
Focal tumor cells stain with myogenin.
Physical examination of the patient at 10 months after surgery revealed a well-healed incision with a palpable skull defect. Magnetic resonance imaging revealed no evidence of recurrent tumor. A cranioplasty is planned after the patient is 3 years of age.
Slides from an additional case of MNTI from Columbus Children's Hospital were obtained; the patient was 3 months old with a 3-week history of a left mouth mass. The patient underwent left partial maxillectomy. The diagnosis was MNTI of the maxilla. The mass was completely excised. The myogenin stain showed scattered positive cells.
DISCUSSION
Based on histological, immunohistochemical, ultrastructural, and biochemical data, MNTI is derived from neural crest cells analogous to developing peripheral nerve and melanocytes. The tumor is composed of melanin-producing epithelial cells and neuroblastic cells. Some patients have elevated levels of vanillylmandelic and homovanillic acid in the urine [2]. The tumor cells usually stain with neural markers, such as neuron-specific enolase, synaptophysin, and CD57. S-100 positivity has been reported in a few cases [3]. MNTIs also stain positively with HMB45 and cytokeratin. A few cases of MNTI appear to be polyphenotypic, as shown by expression of epithelial membrane antigen, GFAP, muscle-specific actin, and desmin [2]. Our current myogenin-positive cases extend the range of unexpected cell marker expression that may be encountered in MNTI and confirm the myogenic potential of these tumors.
Myogenin is a transcriptional regulator found in normal fetal muscle and expressed by rhabdomyosarcoma. Previous studies have shown that myogenin is not a specific marker for rhabdomyosarcoma but can be present in other tumors with myogenous differentiation, such as Wilms' tumors and ectomesenchyomas [4]. Staining in desmoid tumor, infantile myofibromatosis, infantile fibrosarcoma, and synovial sarcoma has also been rarely reported [5].
Skeletal muscle differentiation has been observed in a large number of neoplasms. Germ cell tumors, pleuropulmonary blastoma, medulloblastoma, hepatoblastoma, Triton tumor, and carcinosarcomas are examples. A recent article by Eusebi and colleagues [6] described 3 cases of small-cell neuroendocrine carcinoma with skeletal muscle differentiation. These cases showed focal myogenin positivity. Because of numerous normal and pathologic conditions in which skeletal muscle cells are associated with neural structures and the fact that skeletal muscle differentiation has been induced in normal and neoplastic cell lines, Eusebi and colleagues suggested the existence of “ectomesenchyme.” They postulated that if a program exists and is normally activated in the neural crest of the head and neck region, it is likely that it could be derepressed in abnormal conditions, such as primitive neural tumors [6]. Our cases of MNTI would provide further evidence that such a program exists.
It is well known that neural crest cells are multipotent cells. Cranial neural crest cells have the ability to differentiate into cartilage, bone, connective tissue, pigment cells, and sensory and parasympathetic ganglia. Neural crest cells also exhibit a limited capacity for self renewal. It has been suggested that premigratory neural crest cells in the neural tube may represent neural crest stem cells with the ability to form peripheral nervous system, central nervous system, and non-neuronal derivatives [7]. Interestingly, 3 tumors thought to be of neural crest origin—malignant triton tumor, ectomesenchymoma, and medullomyoblastoma—contain both neural and myogenous components. Malignant triton tumors are composed of malignant peripheral nerve sheath tumor and rhabdomyoblasts. Ectomesenchymomas are usually composed of ganglion cells and rhabdomyosarcoma. Medullomyoblastomas are composed of small undifferentiated cells with myoblasts or myocytes. Occasional primitive neuroectodermal tumors stain with desmin.
Skeletal muscle differentiation is dependent on 4 transcription factors (Myf5, MyoD, myogenin, and Mrf4) known as myogenic regulatory factors. Myf5 and MyoD are required for determination of myoblasts; myogenin and Mrf4 act later as differentiation factors. In a study by Delfini and Duprez [8], ectopic mouse Myf5 and MyoD induced ectopic skeletal muscle differentiation in the neural tubes of chick embryos. Myogenic regulatory factors have the ability to convert various cell types into differentiation-competent myogenic cells.
MNTI has morphologic and immunophenotypic features shared with the developing retina. After formation of the optic cup, the outer wall becomes highly pigmented during week 5 of gestation and forms the pigmented epithelium of the retina. Cells of the inner wall of the optic cup differentiate into neural elements. The pigmented epithelium in both the retina and MNTI synthesizes melanin and expresses immunophenotypic evidence of epithelial differentiation [2]. A search of the literature revealed no reports of muscle differentiation in primary retinoblastomas. However, Dickman and colleagues [9] reported 3 patients with retinoblastoma who showed muscle differentiation in their recurrent tumors.
MNTI is a tumor composed of primitive neuroepithelial cells with polyphenotypic differentiation along neural, melanogenic, and epithelial lines with occasional myogenic, glial, or ganglionic [10] differentiation. Unlike other primitive neuroectodermal tumors, such as neuroblastoma, primitive neuroectodermal tumor/Ewing's sarcoma, retinoblastoma, and medulloblastoma, the majority of MNTIs are not aggressive tumors. Only a small percentage of MNTIs show malignant behavior and those that do lack features predicting recurrence or metastasis [2].
In summary, we report a case of MNTI involving the skull of an 8-month-old boy who showed evidence of primitive myogenic differentiation, as evidenced by myogenin expression. To our knowledge, this is the 1st case report of MNTI with myogenin positivity. An additional case from Columbus Children's Hospital was also focally positive for myogenin. The biphasic histologic appearance of MNTI is diagnostic, but small biopsies might be confused with neuroblastoma, melanoma, rhabdomyosarcoma, and primitive neuroectodermal tumors of the peripheral and central nervous system. Because of the relative benign nature of these lesions, careful deliberation and clinicopathologic correlation is thus advised when confronted with limited biopsies of small-cell tumors with myogenic and neural features.