
Abstract
A case of primary alveolar soft part sarcoma (ASPS) of the heart is reported in an 11-year-old female as 1 of 16 cases of ASPS presenting in the first 2 decades of life in our institutional 17-year review period. The classic alveolar or organoid pattern was inconspicuous as compared to a more diffuse or formless pattern consisting of a population of uniform round cells with abundant eosinophilic cytoplasm, but in addition there was a second, minor population of gigantiform tumor cells with a variety of unusual shapes. Scattered tumor cells contained dense eosinophilic condensations in the cytoplasm. Other unusual features for ASPS in our case included a lymphohistocytic reaction and zonal necrosis. Immunohistochemistry revealed nuclear reactivity for TFE3, and the ASPL-TFE3 fusion transcript was identified by reverse-transcriptase polymerase chain reaction. The only other examples of ASPS involving the heart were 3 cases in the literature of metastatic disease from tumors arising in the soft tissues. This initial case of primary cardiac ASPS joins the list of other types of sarcomas in children that have been reported as primary neoplasms of the heart.
INTRODUCTION
The convergence of an uncommon clinical event, a primary sarcoma of the heart, and an equally infrequent type of soft tissue sarcoma, alveolar soft part sarcoma (ASPS), is reported in an 11-year-old female. This neoplasm also highlighted the fact that APSP does not always have the classic alveolar or organized pattern. Although this tumor had several unusual histologic features, nevertheless the immunophenotype and, more importantly, molecular genetic analysis were characteristic of ASPS.
To the best of our knowledge, this case of an ASPS presenting as a primary neoplasm in the heart is a unique event, as ascertained from our inclusive survey of PubMed with the search terms “heart,” “cardiac,” and “alveolar soft part sarcoma” and “sarcoma” with “heart” and “cardiac.” Several clinical and pathologic series of ASPSs consisting of children alone or adults and children were also reviewed, and no examples of primary or secondary cardiac ASPSs were identified. The only 3 cases of ASPS involving the heart were examples of metastatic disease, which in itself is seemingly a rare event [1–3].
Alveolar soft part sarcoma has been described in a multiplicity of locations but the soft tissues of the lower extremity and the trunk-abdomen are the most common sites of presentation [4–9]. There is also a predilection for the head and neck region, especially in the first decade of life [9]. Most cases are diagnosed in the first 3 decades of life (65% to 70% of cases) so that our case is representative of the age group at presentation and reflects the female predilection that has been documented in larger series [4,5,8].
CASE REPORT
Clinical history
A previously healthy 11-year-old girl presented with fever, cough, and shortness of breath; she was diagnosed with streptococcal pharyngitis and responded to antibiotic therapy. However, a chest roentgenograph to further evaluate the shortness of breath revealed a shadow in relation to the heart that was initially considered a “fat pad”; however, the subsequent echocardiogram revealed a mass in the right atrium. Computed tomography demonstrated a tumor that originated from the right atrium, extended through the pericardium, and invaded the adjacent right lung (Fig. 1). Further diagnostic studies and noninvasive staging showed no evidence of masses elsewhere in central or peripheral body sites.
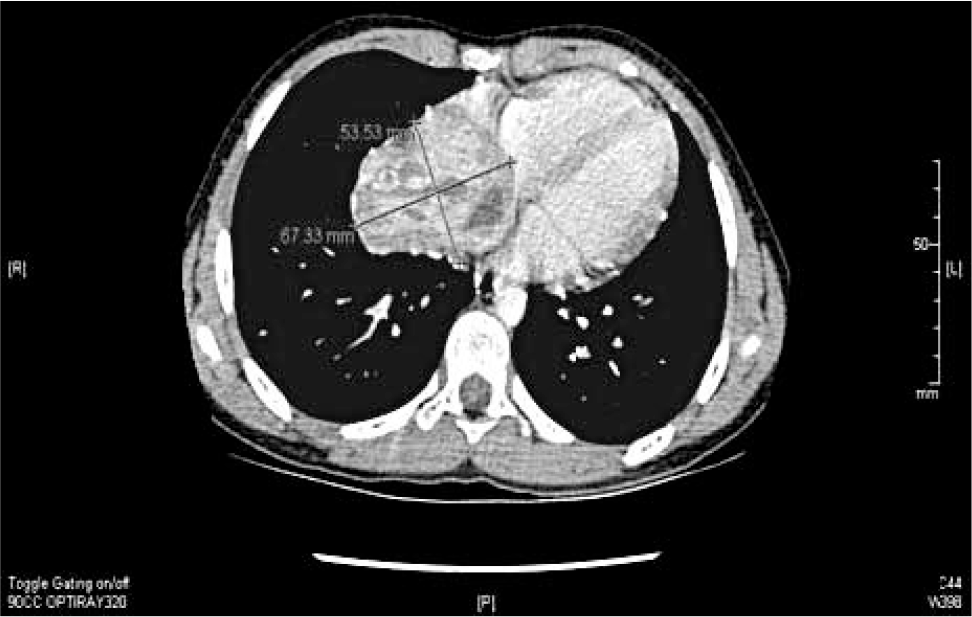
Computed tomography showing a mass filling the right atrium.
At the time of surgery, it appeared that the tumor originated in the right atrium and extended through the right pericardium anterior to the phrenic nerve. The tumor filled virtually the entire right atrial chamber, with resulting obstruction of blood flow from the superior vena cava to the tricuspid valve. After it was determined that the tumor was confined to the heart and pericardium, an excision was performed but it was known at the time that the excision was incomplete posteriorly in order to preserve the phrenic nerve. The resulting cardiac defect was repaired with a pericardial patch.
The patient's post-operative course was uneventful, and she remains tumor-free at 23 months post-excision.
Pathologic findings
Grossly, the tumor consisted of a partially encapsulated rubbery mass, measuring 7.5 × 6 × 5 cm and weighing 84 g. The cut surface had a uniform pale grey appearance with focal hemorrhage.
A representative microscopic field was composed of formless sheets of uniform polygonal cells with abundant eosinophilic cytoplasm and a central nucleus with a single nucleolus and vesicular chromatin (Fig. 2A). A well developed organoid pattern was inconspicuous throughout. Some residual myocardium was identified in the midst of the neoplasm, which was composed of variably sized round cells with abundant eosinophilic cytoplasm. Mitotic figures and anaplasia were inapparent. The brightly acidophilic cytoplasm was uniform from one cell to another except for those cells with elongated crystalline or condensation bodies with a deeper eosinophilic hue than the surrounding cytoplasm (Fig. 2B). Interspersed among the smaller, uniformly sized tumor cells were those larger rounded tumor cells that tended to dwarf the surrounding cells (Fig. 2C). Other somewhat bizarre appearing tumor cells had rectangular and rhomboid shapes (Fig. 2D). Many areas consisted of loosely arrayed tumor cells without a hint of an organoid pattern (Fig. 2C,D). An active inflammatory infiltrate was noted throughout the tumor consisting of lymphocytes, plasma cells, and macrophages among tumor cells (Fig. 2D,E). There were also multifocal areas of coagulative tumor necrosis with surrounding palisades of macrophages (Fig. 2F).
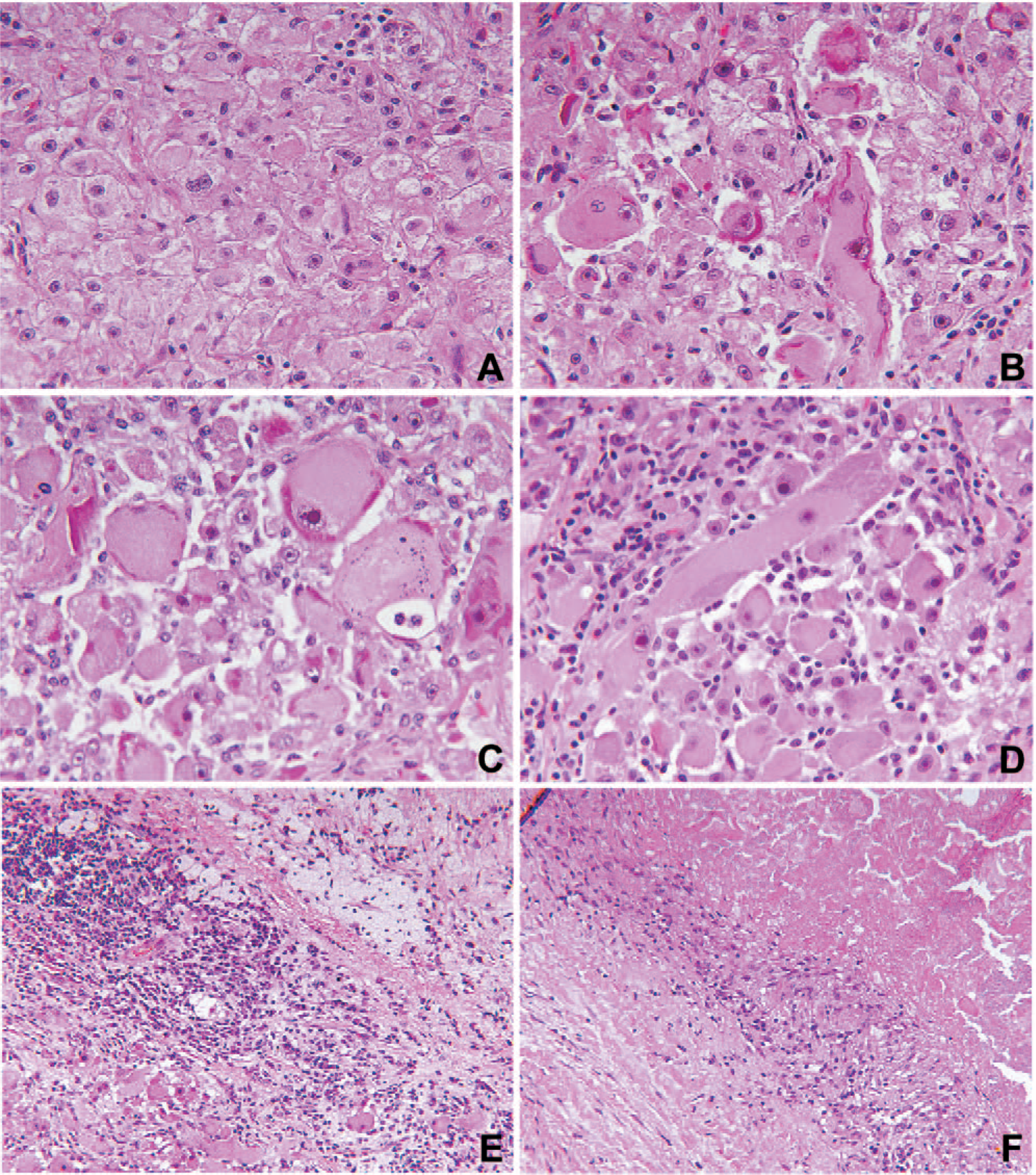
Alveolar soft part sarcoma in the myocardium with
Immunohistochemical stains performed by standard technique–demonstrated focal reactivity for vimentin (V9, Biogenex, San Ramon, CA, USA, 1:2000), smooth muscle actin (IA4, Dako Cytomation, Carpinteria, CA, USA, 1:200) and desmin (33, Biogenex, 1:50) [10] (Fig. 3A–C). The tumor cells were nonreactive for epithelial membrane antigen (E29, Dako Cytomation, 1:2000) and HMB-45 (HMB45, Dako Cytomation, 1:500). There was low-intensity nuclear reactivity for TFE3 (Fig. 3D).
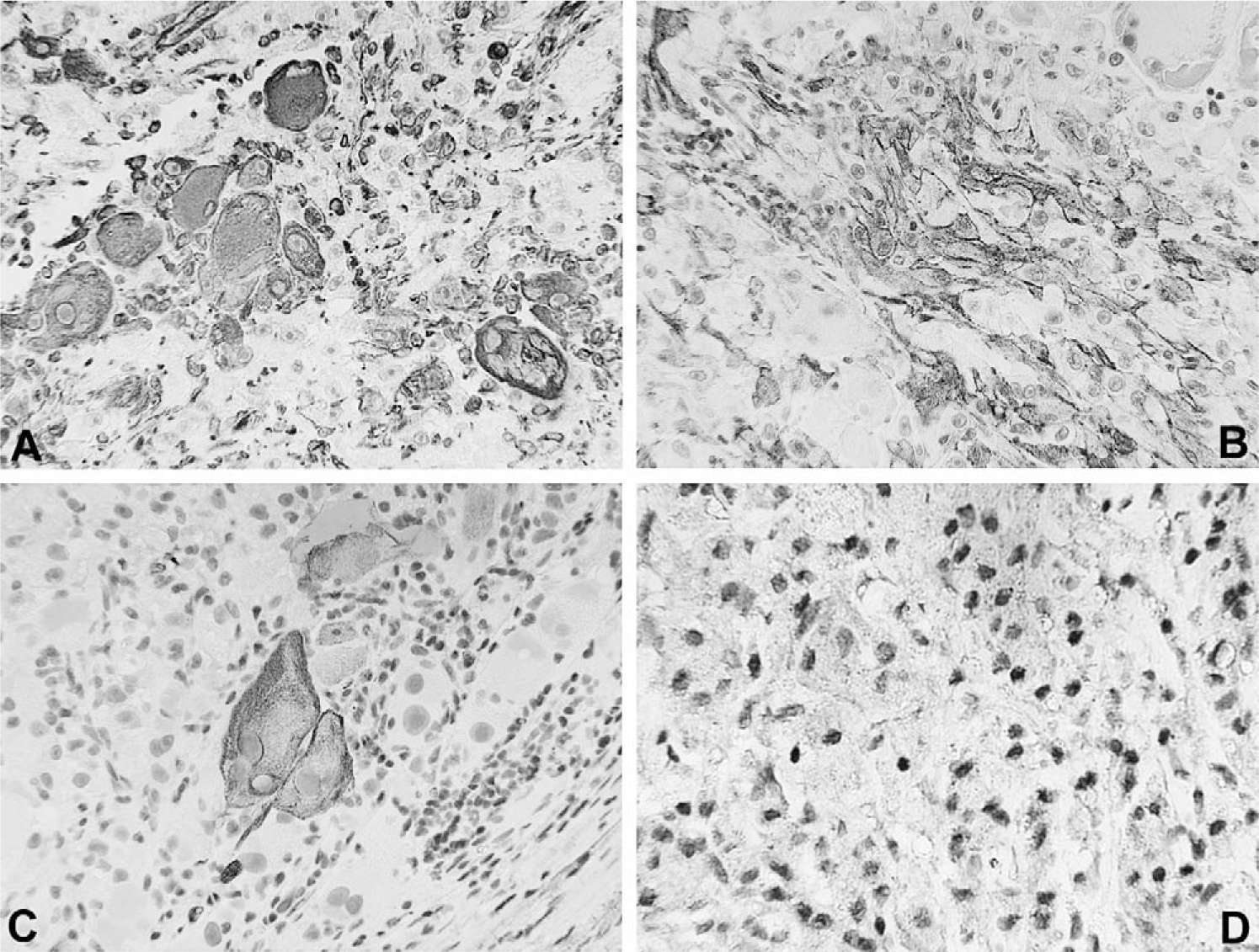
Immunohistochemical staining showing
Reverse-transcriptase polymerase chain reaction (RT-PCR) testing performed by standard techniques demonstrated the ASPL-TFE3 fusion transcript characteristic of ASPS [11] (Fig. 4).
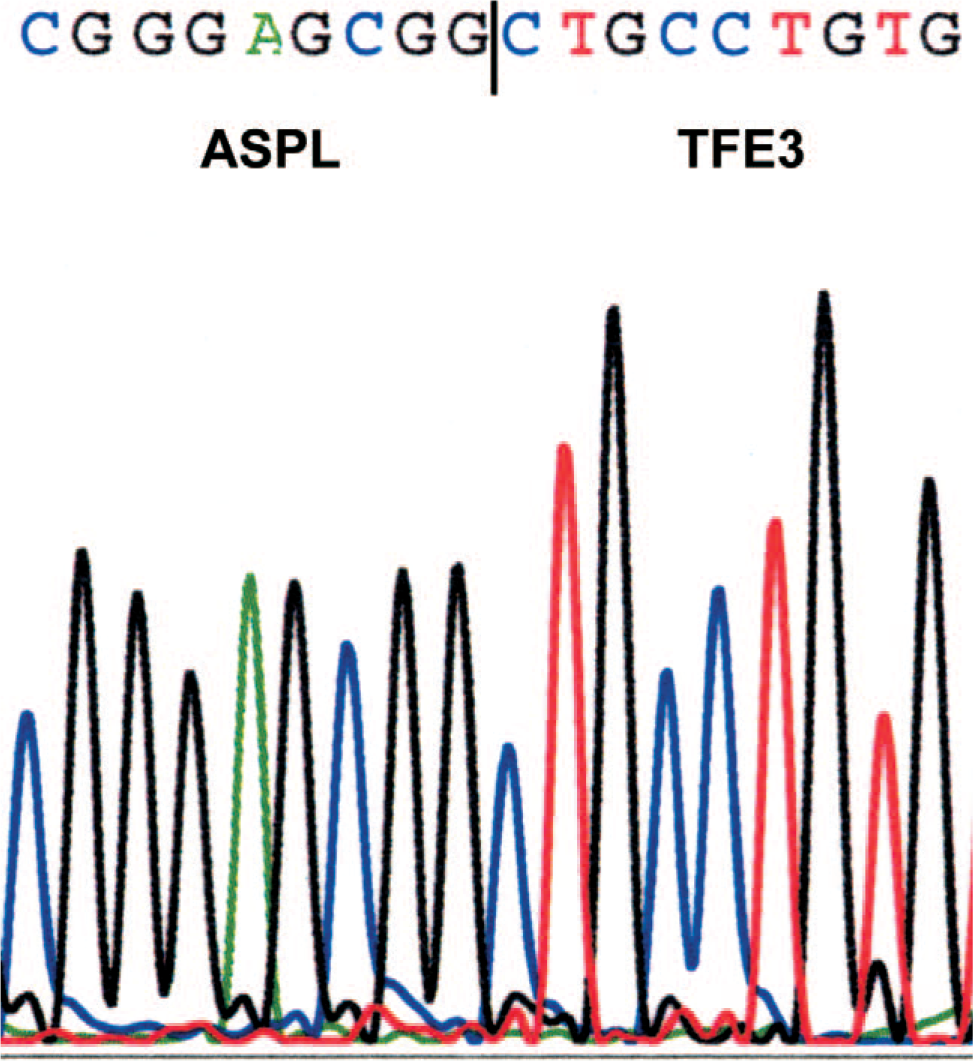
Partial nucleotide sequence of the ASPL-TFE3 fusion transcript. The vertical line indicates the boundary between the region derived from ASPL and the region derived from TFE3 beginning with exon 4, the boundary characteristic of so-called type 1 fusions. A color version of this figure is available in the online journal.
DISCUSSION
The dilemma about the histogenesis and classification of the ASPS began upon its initial description in 1952 more than 50 years ago and has not abated with time [12]. Although there is no consensus about the nature of the ASPS other than a “tumor of uncertain histogenesis,” a great deal has been learned about its morphology, immunophenotype, and cytogenetic attributes [5,13–15]. Even though the ASPS is a rare soft tissue neoplasm, it has been studied extensively as evidenced by the approximately 400 citations with “alveolar soft part sarcoma” in the title dating from 1953 to 2006 in the PubMed data base.
Alveolar soft part sarcoma accounts for approximately 5% of all non-rhabdomyosarcomatous soft tissue sarcomas (STS) in children [16]. It represents only 1% of all STSs in individuals 20 years old or less at diagnosis [17]. One of the largest institutional experiences of 70 cases reported that almost 60% of patients were 30 years or less at the time of diagnosis [4]. Our more modest series of 22 cases consisted of 16 individuals 20 years old or less at diagnosis with mean and median ages of 11 and 14 years old, respectively (Table 1). Our youngest case was in a 1-month-old female with a mass in the thigh. Seven of 16 cases (44%) presented with a tumor mass in the head and neck region. Three of the seven ASPSs were located in the base of the tongue, a site well documented in the report of Fanburg-Smith and associates [18]. Just as the subject of this report, more than 90% of our cases were seen in consultation by one of the authors (L.P.D.). There was more than twice the number of females (11 cases) than males (5 cases). The female preponderance is not limited to our cases, but has been documented by others [7,8]. Bu and Bernstein have offered their explanation for the female predilection as noninactivation of the ASPS gene on the second X chromosome [19].
Alveolar soft part sarcomas presenting in the first 2 decades of life from the files of the Lauren V Ackerman Laboratory of Surgical Pathology, Barnes-Jewish and St Louis Children's Hospitals (1989–2006)

M indicates male; F, female.
Stated age at diagnosis with a mean age of 11 years (range: 1 month to 19 years); median age, 14 years.
Eleven females and 5 males.
The subject of the current report is, to our knowledge, the initial documentation of a primary cardiac ASPS, but there are only 3 reported examples of metastatic ASPS to the heart: in a 42-year-old female with metastases to the right and left ventricles, a 22-year-old female who developed a metastasis in the right ventricle 5 years after the resection of an ASPS in the thigh, and a 13-year-old female with a primary tumor in the forearm [1–3]. Our patient had thorough preoperative imaging studies, and the only mass detected was in the right atrium. Although our patient remains tumor-free since the surgery 23 months ago, ASPL is known for its capacity for delayed recurrences or metastases 5 or more disease-free years after initial diagnosis and treatment [4,5,7,8].
Alveolar soft part sarcoma has been thoroughly studied, in terms of its pathology and immunophenotype, but our case illustrates that not all such neoplasms have the classic organoid pattern nor a uniformly consistent phenotype [5,6]. Evans some years ago was one of the first to draw attention to the “atypical” histologic features, which included nuclear hyperchromatism, pleomorphism, vague nesting or “alveolar” pattern, an attenuated eosinophilic cytoplasm, and increased mitotic activity [20]. More recently, Jong and associates described 2 examples of ASPS, one of which had pseudoglandular profiles and the other had a spindle cell pattern [21]. Our case had several uncommon and even unusual features, including a diffuse, predominantly nonalveolar or nonorganoid pattern of formless sheets of cells. In fact, several of our other ASPSs seen in consultation also lacked the classic organoid pattern. In this respect, the present case would seem to be an example of an ASPS whose “nest-like pattern is inconspicuous or absent entirely” as noted by Weiss and Goldblum [6]. Another unusual finding in our case was the population of “large,” almost gigantiform cells, round cells whose sizes exceeded by several-fold the predominant cell type. Not quite as numerous as the “large rounded cells,” a second population of equally large cells had rectangular or rhomboid shapes. Many tumor cells contained bright eosinophilic cytoplasm condensations. A prominent inflammatory infiltrate and focal necrosis were present, additionally unusual features in our experience with ASPS.
The immunophenotype of the ASPS is not consistent from one case to another including the present one in which only a minority of the tumor cells expressed vimentin, smooth muscle actin, and desmin, whereas the majority of cells were nonreactive for these and several other markers. Because the ASPS can express various myogenic proteins, the suggestion has been made that it is a variant or subtype of rhabdomyosarcoma [22,23]. The latter theory has been subverted in part by the inability to demonstrate MyoD1 expression on a consistent basis [24–26]. However, the molecular genetics of the ASPS would suggest an even more provocative histogenetic relationship to a variant of renal cell carcinoma, which is seen almost exclusively in children [27,28].
A common denominator of the ASPS, irrespective of clinical, pathologic, and immunophenotypic variables, is the unbalanced translocation der(17)t(x;17)(p11;q25) [14,29]. The nonreciprocal translocation produces an ASPL-TFE3 fusion gene that encodes a chimeric protein in which the N-terminal region of ASPL is linked to the basic loop-helix-loop leucine zipper DNA-binding and multimerization domains of TFE3. Consequently, ASPS overexpresses the C-terminal portion of the TFE3 protein, and so immunohistochemical reactivity for nuclear TFE3 is a sensitive and specific ancillary finding as in our case and is supportive of the diagnosis of ASPS in the absence of conventional cytogenetic analysis or molecular testing [30]. It is noteworthy that the unbalanced nature of the translocation in ASPS results in a loss of 17q25 sequences telomeric to ASPL and a gain of Xp11 sequences telomeric to TFE3, although whether the resulting gains or losses in copy number of genes in these regions contribute to the pathogenesis of ASPS is unknown. It is interesting, however, that a balanced translocation t(X;17)(p11;q25) that involves ASPL and TFE3 is found in a type of renal cell carcinoma that is seen almost exclusively in children and young adults [27,28].
Despite the presence of a recurring underlying genetic abnormality, the histogenesis of ASPS is no more certain today than it was more than 50 years ago [31]. The demonstration by electron microscopy that the characteristic periodic acid-schiff–positive cytoplasmic crystals are composed of monocarboxylate transporter 1 and CD147 has also failed to provide any insight into the progenitorship of this neoplasm [32]. So we are left to our own imagination to link some progenitor cell in the kidney to one in the soft tissues and any one of a number of anatomic sites including the heart in which ASPS has been reported.
The present case also contributes to the abbreviated list of primary sarcomas and other neoplasms of the heart in children [33]. Virtually the entire spectrum of STS has been reported as primary cardiac neoplasms, including synovial sarcoma and primitive neuroectodermal tumor as two examples [34,35]. However, angiosarcoma, rhabdomyosarcoma, and leiomyosarcoma are among the more common pathologic types [34]. In children, the majority (75% to 80%) of primary neoplasms of the heart are benign and represented by the rhabdomyoma, typically in the setting of tuberous sclerosis, and the fibroma (myofibroma) [33,35]. In terms of borderline or overtly malignant neoplasms of the heart, we have seen examples of the inflammatory myofibroblastic tumor and rhabdomyosarcoma. Most malignant tumors involving the heart in children are metastatic in nature and include Wilms tumor, alveolar rhabdomyosarcoma, hepatoblastoma, and non-Hodgkin lymphoma. After the diagnosis of ASPS was established in our case, clinical attention was turned to the possibility of a metastasis to the heart from an occult primary site but elsewhere. A thorough evaluation at the time of diagnosis and subsequent follow-up has led to the conclusion that the heart was and remains the only site of disease.