
Abstract
Lung morphogenesis requires the integration of multiple regulatory factors, which results in a functional air-blood interface required for gas exchange at birth. The respiratory tract is composed of endodermally derived epithelium surrounded by cells of mesodermal origin. Inductive signaling between these 2 tissue compartments plays a critical role in formation and differentiation of the lung, which is mediated by evolutionarily conserved signaling families used reiteratively during lung formation, including the fibroblast growth factor, hedgehog, retinoic acid, bone morphogenetic protein, and Wnt signaling pathways. Cells coordinate their response to these signaling proteins largely through transcription factors, which determine respiratory cell fate and pattern formation via the activation and repression of downstream target genes. Gain- and loss-of-function studies in null mutant and transgenic mice models have greatly facilitated the identification and hierarchical classification of these molecular programs. In this review, we highlight select molecular events that drive key phases of pulmonary development, including specification of a lung cell fate, primary lung bud formation, tracheoesophageal septation, branching morphogenesis, and proximal-distal epithelial patterning. Understanding the genetic pathways that regulate respiratory tract development is essential to provide insight into the pathogenesis of congenital anomalies and to develop innovative strategies to treat inherited and acquired lung disease.
LANDMARKS IN LUNG DEVELOPMENT
During gestation, the fetal lung undergoes significant morphological changes to provide an organ capable of maintaining respiration and gas exchange at birth. Lung development initiates with establishment of a lung cell fate from the ventral foregut endoderm [1]. Subsequently, the laryngotracheal groove arises from the floor of the foregut as a ventral outpouching. The proximal portion of this structure gives rise to the larynx and trachea, whereas the distal portion gives rise to 2 ventrolateral buds that will form the left and right primary bronchi. Division of the foregut by a longitudinal septum separates the trachea and dorsal esophagus. The bronchial buds grow into the adjacent splanchnic mesoderm and undergo reiterative branching to give rise to the future respiratory tree. Although lung development is a continuous process, 5 developmental stages have been delineated based on anatomic and histological characteristics [2] (Fig. 1). Functionally, the embryonic and pseudoglandular stages elaborate the conducting airways of the lung; the later canalicular, saccular, and alveolar stages are characterized by vascularization and reduction of mesenchyme to form a thin air-blood interface. There is a continuing complex process of lung growth postnatally, resulting in rapid expansion of the gas exchange surface area. Concomitant with lung morphogenesis is cell differentiation within the respective tissue compartments. The lung is lined by endodermally derived epithelium, which differentiates into several specialized cell types, including ciliated, basal, Clara, and goblet cells in the proximal airways, and type I and type II cells in the distal alveoli [3]. Mesenchyme originating from splanchnic mesoderm gives rise to the vascular network, smooth muscle, cartilage, and other connective tissue elements that develop in parallel and surround the airways [4].
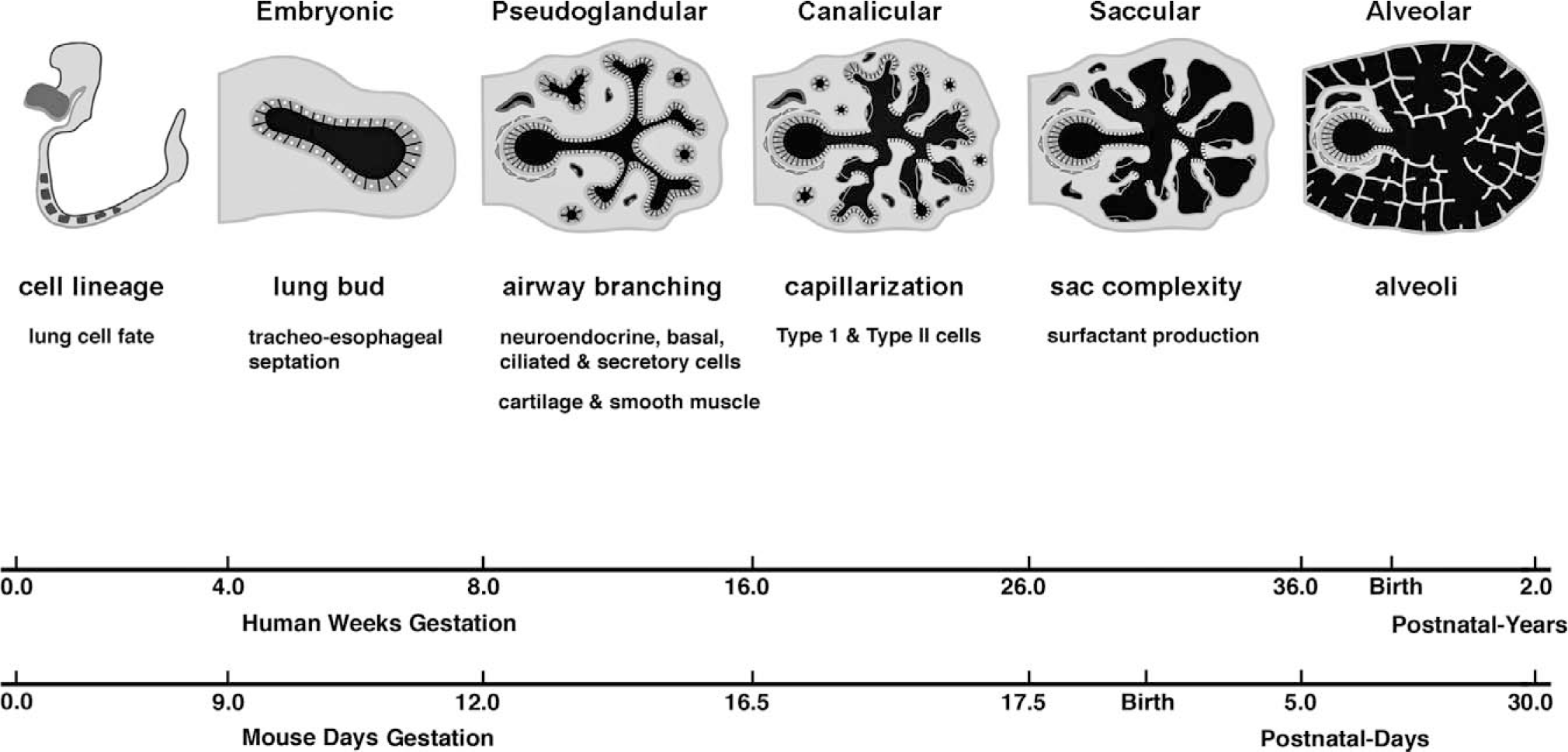
Timeline of lung development. Morphological stages and major events in the developing human and mouse lung.
Development of the respiratory tract thus requires a coordinated sequence of processes, which when altered could disrupt key phases of lung development, including specification, growth, and differentiation. Gene ablation in mice (through direct disruption of a genetic locus or conditional gene targeting) and embryonic and lung explant assays have permitted identification of factors required for distinct phases in lung development. The recurrent theme emerging from these studies is that lung development results from successive epithelial-mesenchymal interactions. Growth factor signaling and induction of responsive transcription factors orchestrate this interplay between developing epithelial and mesenchymal structures by influencing cell fate, proliferation, migration, and differentiation. In the subsequent sections, we highlight molecular mechanisms that regulate fundamental aspects of lung development, including specification of a lung cell fate, primary lung bud induction, tracheoesophageal septation, branching morphogenesis, and proximal-distal epithelial differentiation. Cellular and transcriptional programs mediating alveolarization and injury and repair of the lung are not discussed in this review [5]. Molecular regulation of pulmonary vascular development was recently covered in this journal by Galambos and DeMello [6], and reviews of the pulmonary neuroendocrine system and surfactant dysfunction disorders are forthcoming.
SPECIFICATION OF A LUNG CELL FATE
The lung shares a common embryological origin with other foregut endoderm derivatives, including the liver, pancreas, thyroid, and gastrointestinal tract. During early somitogenesis, approximately 8.0 days postcoitum in the mouse, the anterior portion of the embryo invaginates to form the foregut, of which the ventral portion ultimately gives rise to the thyroid, thymus, lung, liver, and ventral pancreas [7] (Fig. 2). Development of the lung can first be identified morphologically by mouse embryonic day 9.5 (E9.5) as 2 lateral buds of proliferating endoderm cells present in the ventral foregut. However, before organ bud formation, between E8.5 and E9.0, presumptive organ domains are delineated by the regional expression of tissue-specific genes along the anterior-posterior axis of the gut endoderm [8,9]. For example, Hex is expressed in the thyroid and liver domains, Nkx2.1 (also named thyroid transcription factor 1 [Titf1] and thyroid enhancer binding protein [T/EBP]) in the thyroid and lung domains, and Pdx1 (pancreatic duodenal homeobox 1) in the pancreas domain [10–12]. Although many studies have focused on branching morphogenesis and terminal cell differentiation of the lung, the molecular mechanisms underlying the earliest step of lung development, specification, or induction of a lung cell fate from the foregut endoderm are not well understood. Progenitors of the trachea and lung are first visualized in the prospective lung region of the foregut by the expression of Nkx2.1 at E9.0 (Fig. 4A) [13]. Although the timing of when a respiratory cell fate is induced from the foregut endoderm remains incompletely defined, in vitro studies using mouse embryo endoderm explants indicate that diffusible fibroblast growth factor (FGF) signaling emanating from the neighboring heart (cardiac mesoderm) is essential for lung specification [9]. Cardiac mesoderm and FGF influences the anterior-posterior fate of the ventral foregut endoderm in a dose-dependent manner (Fig. 3). Specifically, endoderm cultured in isolation defaults to a pancreatic fate, whereas endoderm exposed to an increasing amount of cardiac mesoderm or FGF results in successive induction of a liver and then a lung or thyroid fate [9,14,15]. These findings correlate with the temporal activation and spatial domain of lineage-specific markers in vivo as well as the intensity of FGF expressed in the heart. Pdx1 is the earliest cell-specific marker to be activated in the foregut endoderm, and its expression is confined to the ventral lip, distal to the developing heart; in contrast, Nkx2.1 comes on in the prospective lung field a half a day later and close to the heart [9,15].
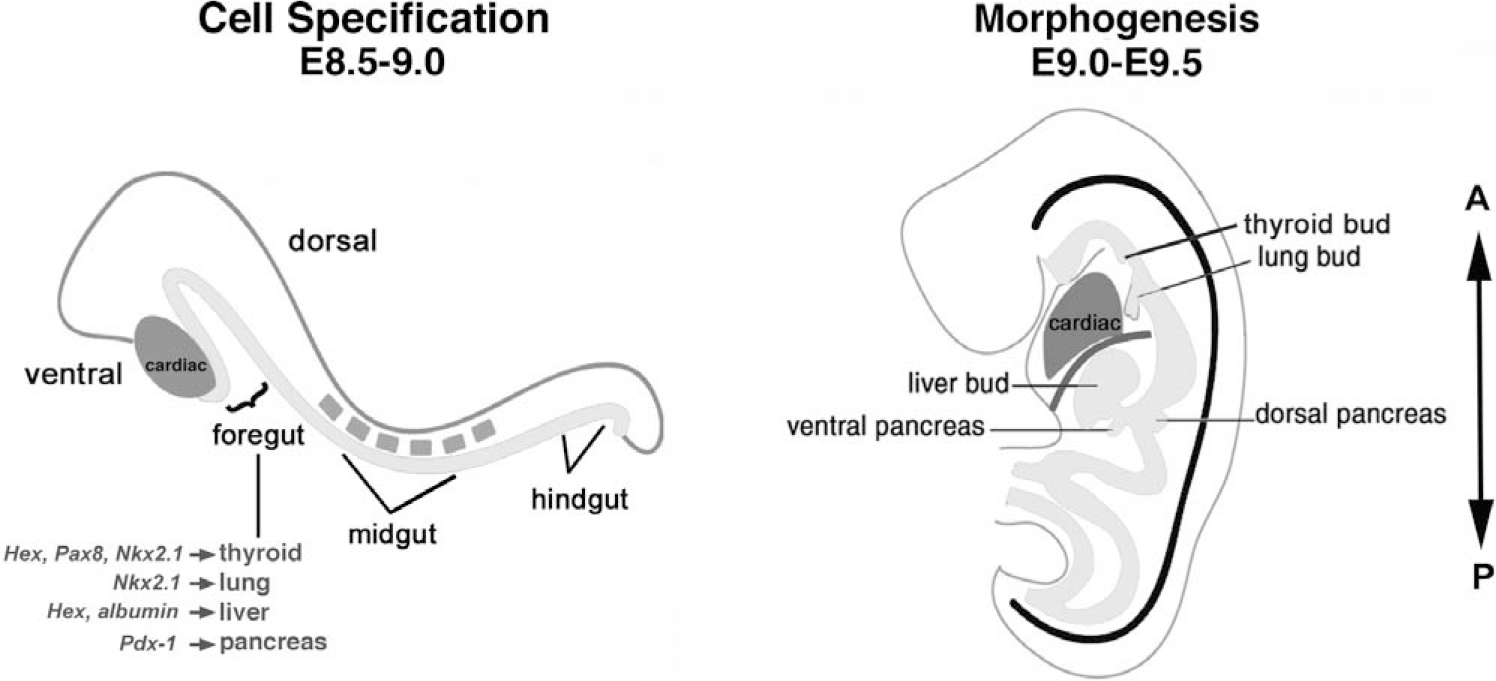
Schematic view of embryos during lung specification and morphogenesis stages. The ventral portion of the foregut endoderm gives rise to different cell lineages, including the lung. The regional expression of tissue-specific genes along the anterior-posterior (A-P) axis of the gut endoderm delineates organ-specific domains, well before bud formation at the morphogenesis stage of development.
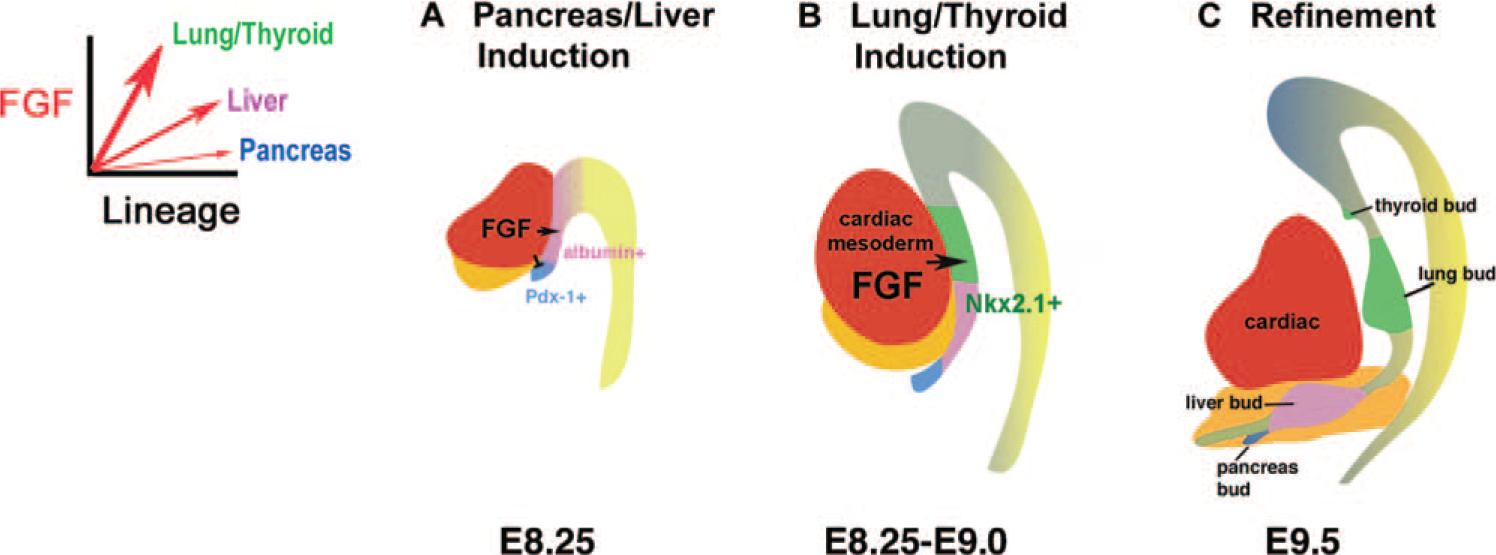
Proposed model of ventral foregut specification. An increasing threshold of fibroblast growth factor signaling from the heart (red) results in successive induction of a pancreas (blue), liver (pink), and lung/thyroid (green) cell fate from the adjacent foregut endoderm.
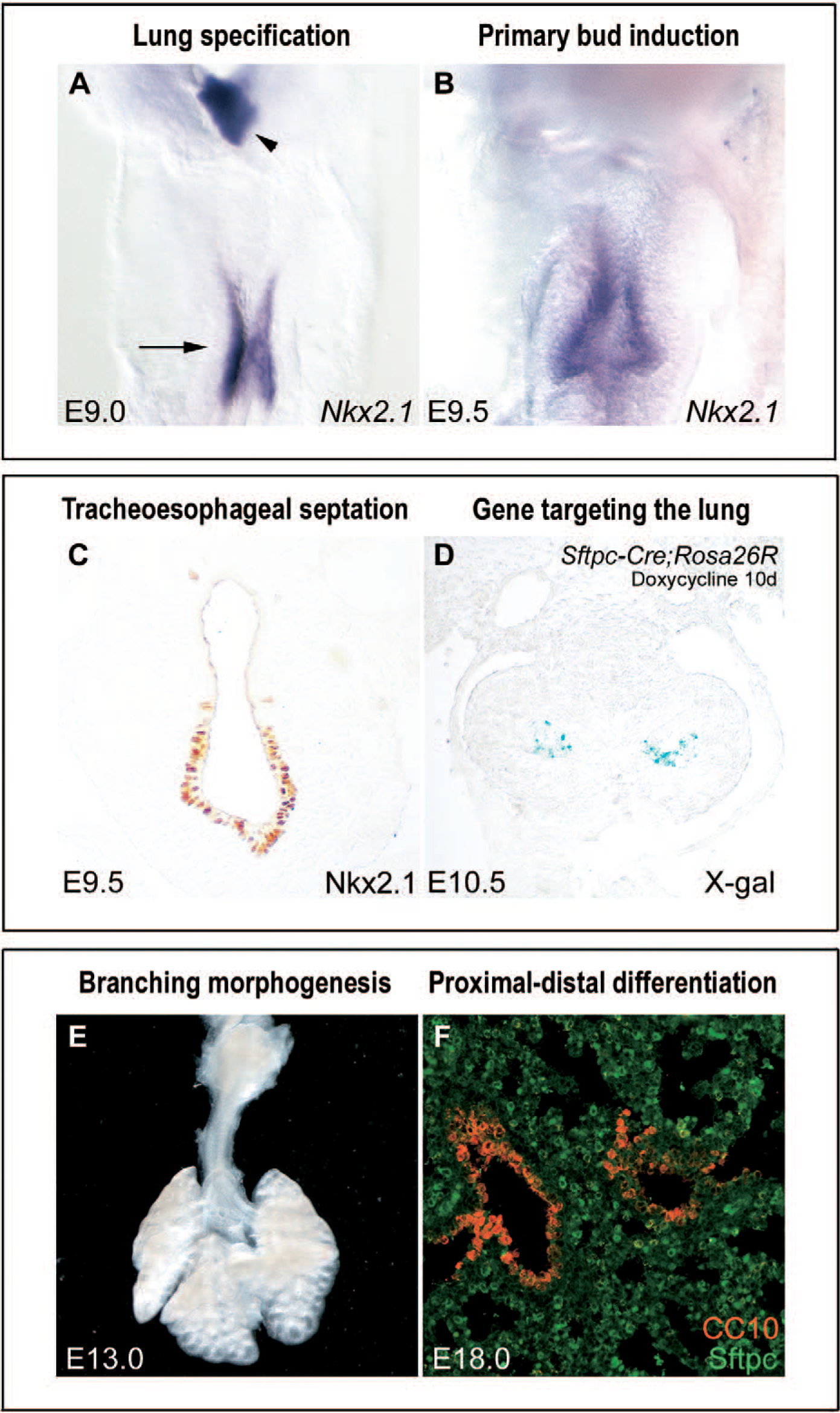
Key phases of early mouse lung development. (A) Lung and tracheal progenitors (arrow) are identified at embryonic day 9.0 (E9.0) by the expression of Nkx2.1 (purple), which also denotes the thyroid (arrowhead). (B) At E9.5, the right and left lung buds arise from the ventral-lateral aspect of the foregut, invading into the adjacent mesoderm. (C) Concurrent with primary lung bud formation, the tracheal primordium forms from the ventral aspect of the foregut, separating from the dorsal foregut/primitive esophagus by a tracheoesophageal septum. (D) The 3.7-kb human Sftpc promoter is expressed at high levels in the developing and mature lung and has been used to drive ectopic expression of many genes of interest (here the reporter gene beta-galactosidase blue staining) into the early respiratory epithelium [112] (Sftpc-Cre mice provided by Dr. Anne-Karina Perl and Dr. Jeffrey Whitsett, Cincinnati Children's Hospital Medical Center). (E) From E10.5 to E16.5, the epithelium undergoes branching morphogenesis, a reiterative process involving bud outgrowth, elongation, and dichotomous subdivision of terminal units. (F) As this occurs, a proximodistal gradient is established in the developing lung with bronchial and alveolar differentiation, distinguished by CC10 (red) and Sftpc (green) expression, respectively (Sftpc antibody provided by Dr. Susan Wert, Cincinnati Children's Hospital Medical Center).
All foregut derivatives are specified within the domain of Foxa1 and Foxa2 gene expression, transcription factors that play a significant role in foregut differentiation and morphogenesis [16,17]. A null mutation in Foxa2 results in failure of the foregut endoderm to close into a tube; consequently, no organs, including the lung bud, can form [18]. In vitro, Foxa2 activates transcription of the homeodomain gene Nkx2.1 [19] and is thought to cooperate with Nkx2.1 to define cell lineage within the pulmonary epithelium [11,13,20]. Early on, Nkx2.1 is expressed in all pulmonary epithelial cells but becomes progressively restricted to proximal Clara cells and distal type II cells with development [21,22]. Targeted disruption of this gene has demonstrated that while Nkx2.1 plays an essential role in several phases of lung development, including tracheoesophageal septation, branching morphogenesis, and terminal cell differentiation, it is not required for specification [11,13,23]. Mice deficient in Nkx2.1 form lungs, although they are arrested distal to the lobar bronchi and contain only ciliated and mucus-secreting cells, characteristic of proximal lung [11,13,24]. In humans, heterozygous loss of function mutations in the NKX2.1 gene, localized to chromosome 14q, has been described in infants and children with recurrent respiratory distress and thyroid and central nervous system dysfunction [25–27].
To date, a compound null Gli2−/−/Gli3−/− mutation is the only genetic model in which the tracheal and lung anlage fail to arise from the foregut endoderm [28]; the thymus, liver, and pancreas form are but hypoplastic. The Gli family of zinc finger transcription factors has been implicated in transduction of sonic hedgehog (SHH) signal in the endoderm, and Gli1, Gli2, and Gli3 are expressed in the splanchnic and early lung mesenchyme [29,30]. Of interest, this phenotype is much more severe than that seen in Shh- null mutants, who are born with a lung, albeit abnormal [31] (see below). Mice with single mutations in Gli2 or Gli3 have tracheal stenosis and lung lobation and branching defects, similar to those seen in Pallister-Hall syndrome, caused by frame-shift mutations in the Gli3 gene [28,30,32].
PRIMARY LUNG BUD FORMATION
In mice, primary lung buds are evident at E9.5 (Fig. 4B). Akin to specification, lung budding and branching morphogenesis arise from dynamic and reciprocal interactions between the early epithelium and mesenchyme. Soluble factors have long been implicated in branching morphogenesis, since it was discovered that transposition of early-stage distal lung mesenchyme onto the tracheal epithelium can induce supernumerary branching as well as type II cell differentiation with lamellar bodies and surfactant protein C (Sftpc) expression [33]. Studies in Drosophila have determined that primary branching of the lung is spatially regulated by an FGF ligand, the branchless gene [34]. The branchless gene activates an FGF receptor (breathless) in the endoderm to induce tracheal development [35,36]. This FGF signaling pathway appears to be evolutionarily conserved in mammals because FGF10 and its receptor FGFR2b (expressed throughout the respiratory epithelium) are critical for lung bud formation. Fgf10, present at discrete sites in the mesenchyme around the distal lung buds, locally induces and guides bud growth by promoting endodermal proliferation and chemoattraction [37–39]. Deletion of either Fgf10 or Fgfr2b interferes with primary lung budding and peripheral cell differentiation; transgenic lungs are characterized by only a trachea or trachea and 2 bronchi [40–43]. The similar phenotypes of Fgf10- and Fgfr2b-deficient mice suggest that FGF10 from the mesenchyme binds to FGFR2b in the epithelium to activate proliferation and migration of the epithelial cells, leading to directional outgrowth of the lung bud (Fig. 5A).
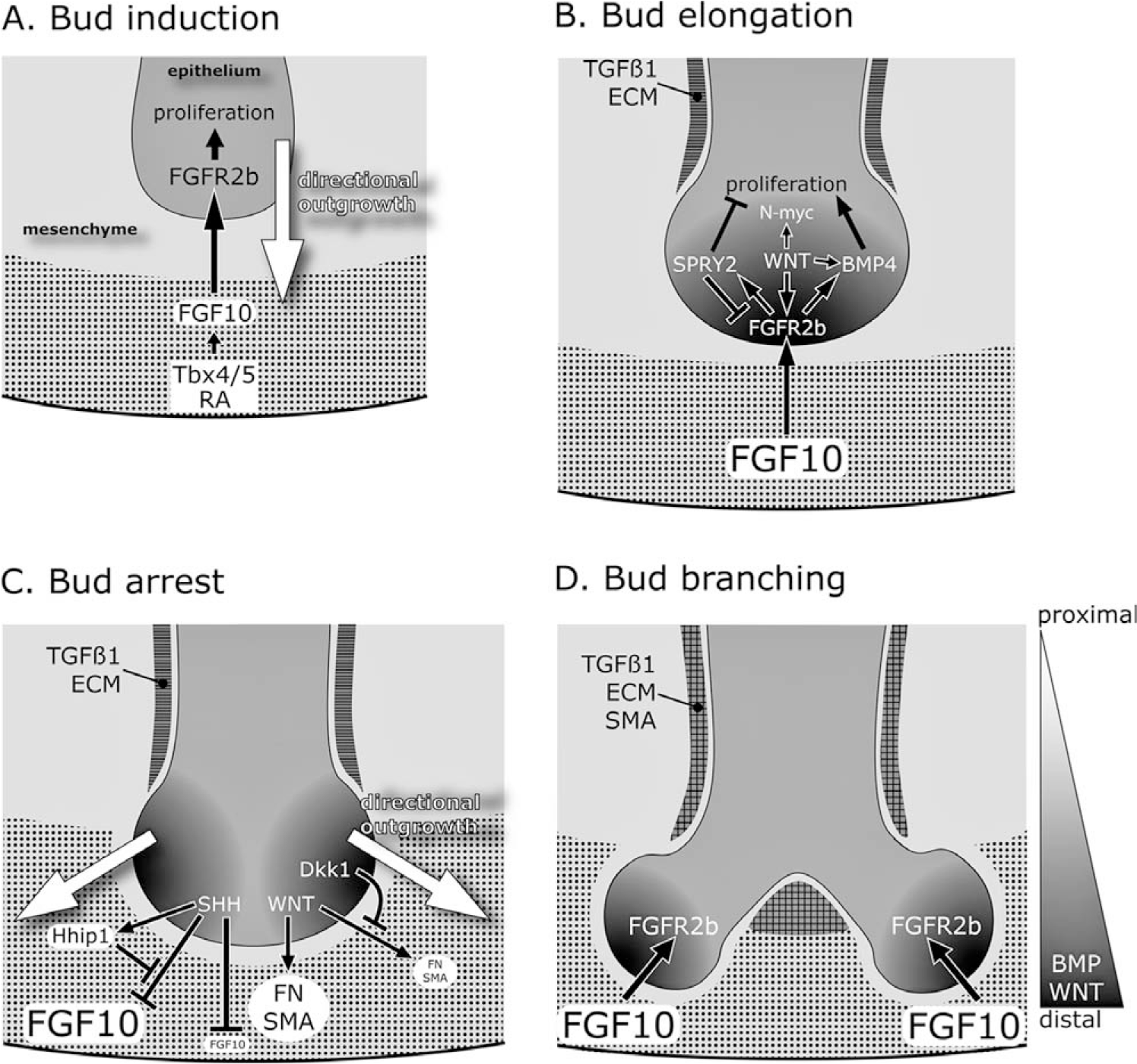
Models of the interactions among key molecules in primary bud formation and branching of the lung. Mesenchyme is depicted in light gray and endodermal epithelium in dark gray. (A) Fgf10 expression (dotted pattern) in the distal mesenchyme is regulated by Tbx genes and retinoic acid. FGF10 diffuses and binds to FGFR2b in the epithelium to induce a bud through proliferation and directional outgrowth (white arrow). (B) As the lung bud elongates, FGF10-FGFR2b signaling in the distal epithelium (black gradient) induces Bmp4 and Spry2. Epithelial WNT signaling also promotes Fgfr2b and Bmp4 expression, which stimulates epithelial proliferation and bud outgrowth. In contrast, SPRY2 controls the size of the lung bud by repressing FGF signaling and proliferation. In the proximal mesenchyme, transforming growth factor-beta1 represses Fgf10 expression and promotes synthesis of extracellular matrix (horizontal striped pattern), including collagen and fibronectin, which might serve to prevent ectopic budding. (C) Elongation of the lung bud arrests because sonic hedgehog (SHH) in the distal epithelium negatively regulates Fgf10. This is counterbalanced by Hhip1, which maintains Fgf10 expression by sequestering the SHH protein. Low SHH levels in the proximal region of the bud allows for higher FGF10-FGFR2b signaling (2 black gradients at the lateral sides) and induction of lateral buds. Lateral bud branching might be facilitated by canonical WNT signaling, which induces fibronectin and alpha-smooth muscle actin in the mesenchyme near the tip of lung bud. Dkk1 inhibits WNT signaling around the distal bud. (D) Resultant secondary lung branches with transforming growth factor-beta1 and extracellular matrix localized to the mesenchyme at the branch point, as well as around the proximal region. A new cycle of bud induction and outgrowth continues from B. As well as their role in branching, WNTs and bone morphogenetic proteins in the distal epithelium influence proximal-distal differentiation of the lung.
Several studies have implicated retinoic acid signaling in regulating Fgf10 expression and bud formation [44,45]. Deficiency of vitamin A or compound null mutations of the retinoic acid receptor genes results in severe abnormalities of the lung, including tracheoesophageal fistula, pulmonary hypoplasia, and agenesis of the left lung [46,47]. In the early lung, retinoic acid synthesis and use are most prominent in the foregut, where the primary lung buds are emerging [48]. Disruption of retinoic acid signaling in E8.5 foregut explants interferes with Fgf10 expression and primary bud formation [44].
T-box (Tbx) transcription factors have also been shown to be positive regulators of Fgf10 expression (Fig. 5A). In chick embryos, Tbx4 and Fgf10 are coexpressed in the foregut mesoderm adjacent to endodermal Nkx2.1 expression in the early lung field. Misexpression of Tbx4 induces ectopic Fgf10 expression and ectopic buds, which express Nkx2.1 [49]. In mouse embryonic lung cultures, repression of Tbx4 and Tbx5 expression through antisense oligonucleotides suppresses Fgf10 expression in the mesenchyme and eliminates formation of new lung branches [50]. Thus, expression of factors in the early foregut mesoderm, including retinoic acid, Fgf10, and Tbx transcription factors are critical for induction of the primary lung bud.
TRACHEOESOPHAGEAL SEPTATION
Concurrent with formation of the primary lung bud is septation of the trachea from the esophagus. The early expression of Nkx2.1 at E9.0 demarcates the dorsoventral boundary of the foregut endoderm, distinguishing the lung primordium from the esophagus [13] (Fig. 4C). This dorsoventral separation is absent or incomplete in mouse embryos with null mutations in Nkx2.1 and Shh, resulting in a tracheoesophageal fistula, as well as lung hypoplasia [13,31,51]. In contrast to Nkx2.1−/− mice, which have a defect in distal lung differentiation, normal proximodistal differentiation of epithelial cells is preserved in Shh−/− mice with expression of both Sftpc and Clara cell protein 10 (CC10) genes. Haploinsufficiency of Foxf1, a Shh target, causes esophageal atresia and tracheoesophageal fistula, which is also present in Gli2−/−/Gli3+/− mice, compound retinoic acid receptor mutants, and Tbx4 deficiency [28,31,49,52,53]. Human malformations of the foregut, including laryngeal clefts, tracheoesophageal fistula, and esophageal and tracheal atresia, occur in isolation or part of more complex syndromes, including VACTERL, Smith-Lemli-Opitz, and Pallister-Hall, the latter a defect in the SHH signaling pathway [54–56]. Although many signaling molecules regulate both tracheal and lung morphogenesis, Fgf10- and Fgfr2-null mutants have normal septation of the foregut, indicating that formation of the proximal and distal respiratory tree may be genetically distinguishable. Lineage analysis in the lung using Cre recombinase to activate a floxed reporter gene under control of the doxycyline-dependent 3.7-kb human Sftpc gene promoter (Fig. 4D) also suggests that the progenitor cells of the trachea and bronchi differ in origin from those that will form the distal airways and alveoli [57].
LEFT-RIGHT ASYMMETRY
In mice and humans, the lung has a left-right asymmetric pattern. Asymmetry of the lung lobes is dependent on early determinants of left-right axis specification that is largely regulated by Tgf-beta–related genes, such as activin receptor II, Lefty1, Lefty II, Nodal, and Pitx2. Of interest, the majority of the left-right determinant genes are expressed in the node and/or lateral plate mesoderm from E8.0 to E8.5, well before the primary lung bud forms at E9.5. In mice, disruption of activin receptor IIB, Foxj1, Pitx2, Pkd2, or Nodal results in right pulmonary isomerism (bilateral 4-lobed lungs) [58–62], whereas inversin- and Lefty1-deficient mice show left pulmonary isomerism (bilateral single lobe) [63,64]. As well as a lung phenotype, there is global asymmetry with cardiac defects and visceral situ inversus.
BRANCHING MORPHOGENESIS
Branching morphogenesis results in formation of the conducting airways down to the terminal bronchioles, a process involving bud outgrowth, elongation, and subdivision of terminal units [65] (Fig. 4E). The size and shape of the lung bud are regulated by both positive and negative signaling between the growing epithelial bud and the mesenchyme. As well as primary lung bud induction (see above), FGF10-FGFR2b signaling controls secondary and subsequent bud formation. FGF7, which shares high-sequence homology to FGF10 and utilizes the same receptor, also influences lung branching by promoting epithelial cell proliferation and expansion [66–69]. Misexpression of FGF7 using the Sftpc or rat Clara cell secretory protein promoter in mice during the pseudoglandular stage alters lung branching with large cystic spaces in the distal lung [70].
Several signaling pathways have been shown to be negative regulators of lung epithelial proliferation, which appear to play a role in counteracting the bud-promoting effect of FGFs. During elongation of the bud, FGF10-FGFR2b signaling induces Sprouty2 (Spry2), as well as bone morphogenetic protein 4 (BMP4) in the distal tip of the epithelium (Fig. 5B) [39,53,71,72]. SPRY family members are inducible negative regulators of FGF signaling, as well as other tyrosine kinase signaling pathways; mSPRY2 inhibits mitogen-activated protein kinase activity in mouse lung epithelial cells in response to FGF10 stimulation [73]. Reduction of Spry2 results in increased branching, cell proliferation, and cell differentiation in cultured embryonic mouse lungs [74]. Conversely, overexpression of Spry2 in the distal epithelium under control of the Sftpc promoter impairs branching with decreased epithelial cell proliferation [72]. Thus, SPRY2 inhibition of FGF10-FGFR2b signaling appears to control the size of the lung bud by limiting its outgrowth.
Bud formation is also controlled by SHH signaling. Shh is highly expressed in the epithelium of the distal lung buds, from where it diffuses to activate signaling in the adjacent mesenchyme through its receptor patched (Ptch1) and the Gli transcription factors [75] (Fig. 5C). Data from lung explants and transgenic mice suggest that the role of SHH signaling is to spatially restrict FGF10 levels in the mesenchyme surrounding the bud tips, thus limiting further bud outgrowth [31,37,76]. In Shh-null mutant mice, Fgf10 is no longer restricted to focal areas but distributes broadly throughout the distal mesenchyme; transgenic lungs are cystic with a defect in branching, and the mesenchyme shows enhanced cell death and decreased cell proliferation [31,51]. In contrast, Fgf10 expression is repressed in lung mesenchyme of transgenic mice that overexpress Shh throughout the epithelium [75]. Overexpression of Shh also leads to hypercellularity of the lung, with excessive mesenchyme between the alveolar spaces [75]. Thus, SHH not only regulates FGF10 expression but also serves as a trophic factor for the lung mesenchyme. SHH induction of the hedgehog interacting protein Hhip1 in the distal mesenchyme may contribute to the regulation of FGF10 expression [77]. Hhip1 attenuates SHH signaling by sequestration of the Hedgehog protein, thus releasing SHH-mediated repression of FGF10 [77] (Fig. 5C). In Hhip1-null animals, branching is severely inhibited because of increased Hedgehog signaling and repression of Fgf10 [78].
The transforming growth factor-beta (TGFβ1) subfamily is also implicated in branching morphogenesis, specifically in regulating epithelial cell proliferation and extracellular matrix deposition [79]. Tgfβ1 is expressed in the mesenchyme and preferentially accumulates around the bronchiolar ducts and airway branch points where collagen, fibronectin, and proteoglycans are also present [80]. Overexpression of Tgfβ1 in organ culture or misexpression of Tgfβ1 in the distal epithelium of transgenic mice has a negative regulatory effect on branching morphogenesis, whereas abrogation of TGFß type II receptor signaling stimulates lung branching with increased epithelial cell proliferation [81,82]. Because TGFß1 has been shown to repress Fgf10 expression in the lung mesenchyme [76], it may be part of the mechanism that prevents budding in the proximal parts of the lung by inhibiting FGF10 and promoting synthesis of extracellular matrix.
Bone morphogenetic proteins are signaling molecules that play multiple roles in lung development. BMP4 is present in the early mesenchyme but is not present in the epithelium until E11–12, where it is localized to the distal lung buds [83–85]. Although there has been some debate on the role of epithelial-derived BMP4 in branching morphogenesis, recent data in which BMP receptor type Ia or BMP4 was disrupted in the epithelium using the Sftpc-Cre transgene indicate that autocrine BMP signaling promotes proliferation and morphogenesis of the distal epithelium [39,84,86] (Fig. 5B).
Functional roles for WNT signaling in lung branching have also been reported. Detection of nuclear localized beta-catenin and Tcf/Lef transcription factors, components of the WNT pathway, can monitor activation of canonical WNT signaling. These factors are increased in the distal lung epithelium at the sites of active branching, as well as in the mesenchyme adjacent to proximal airways where the smooth muscle will form [87,88]. Disruption of canonical WNT signaling in the lung by targeted deletion of beta-catenin or expression of the diffusible WNT inhibitor Dickkopf 1 (Dkk1) prevents distal lung proliferation and differentiation [89,90]. This defect has been linked to failure of Fgfr2b induction in the distal lung epithelium where WNT signaling is inhibited [90] (Fig. 5B). Furthermore, Dkk1-treated lung explants show a reduction in fibronectin and alpha-smooth muscle actin expression in lung mesenchymal cells, as well as impaired vascular development [88] (Fig. 5C). Fibronectin deposition is essential for cleft formation during lung bud branching [91]. In contrast to the above findings, lungs of homozygous Wnt5a-mutant embryos exhibit overbranching of the distal airways; inhibition of lung maturation; and increased expression of Fgf10, Bmp4, and Shh [92]. WNT5a is a noncanonical WNT, normally expressed in the lung mesenchyme and distal epithelium at E12 [92,93]. Further studies are needed to clarify whether there are distinct functions of canonical and noncanonical WNTs.
PROXIMAL-DISTAL PATTERNING AND CELL DIFFERENTIATION
Epithelial cell lineages are arranged in a distinct proximal-distal spatial pattern in the airways, which become morphologically apparent during the pseudoglandular stage [1]. Ciliated, basal, secretory, and neuroendocrine cells are the major cell types constituting the proximal epithelium, whereas type I and type II alveolar cells make up the distal epithelium. The lineage relationships between the different cell types have not been delineated, and the existence and identity of progenitor cells, which may play a role in lung injury and repair, are still under investigation [94]. There is evidence from cell injury models to suggest that basal cells, Clara cells, and type II alveolar cells can function as progenitors, because they have the capacity to proliferate and repopulate the damaged epithelium after acute lung injury [95–100]. Of interest, early on in embryonic development, the undifferentiated epithelium coexpresses several lineage markers, including Sftpa, CC10, and calcitonin gene-related peptide, the latter a marker of neuroendocrine cells [101]. Cells positive for Clara- and type II–specific markers have also been identified in the adult lung in the bronchioalveolar duct region and become more numerous after bleomycin- or naphthalene-induced injury [102,103]. In vitro, these coexpressing cells self-renew and give rise to cells that express markers for Clara, type I, or type II cells, providing the best evidence that multipotential cells in the lung exist and become reenlisted during epithelial repair.
Although ciliated cells have been considered to be terminally differentiated, recent data suggest that these cells can also proliferate and repopulate the epithelium in response to naphthalene injury or partial pneumonectomy [104,105]. However, a genetic lineage–labeling approach using Foxj1-GFP mice provides conflicting data by demonstrating that ciliated cells change their morphology in response to lung injury but do not proliferate, self-renew, or transdifferentiate during repair of the epithelium [94]. The winged helix transcription factor Foxj1 is necessary for ciliated cell development in the lung as well as left-right axis patterning, with mutational abnormalities in the mouse similar to those seen in Kartagener syndrome [106].
Tissue recombination experiments have demonstrated that a proximal versus distal epithelial phenotype is dictated by soluble factors from the adjacent mesenchyme; within a restricted time window, distal lung mesenchyme can reprogram the tracheal epithelium to take on a type II cell phenotype, and, conversely, tracheal mesenchyme can induce proximal differentiation (ciliated and mucous cells) in distal lung epithelium [107,108]. Early studies have also shown that the quantity of mesenchyme is influential in inducing a specific epithelial cell fate [109]. A small amount of recombined mesenchyme is able to direct cell differentiation toward a bronchiolar phenotype; however, an increased amount of the same mesenchyme will induce the epithelial cells to differentiate toward an alveolar phenotype. Thus, an increasing concentration of the same signals or morphogens expressed by the mesenchyme can induce different cell fates in identical epithelial cells.
BMP4 and canonical WNTs both serve as morphogens, which regulate proximal-distal cell fate in the lung along a gradient of signal (Fig. 5D). Inhibition of BMP signaling through transgenic expression of BMP antagonists (noggin, gremlin) in the epithelium promotes proximalization of the lung with ciliated and Clara cells in the peripheral compartment [83,110]. A similar phenotype results from the targeted deletion of beta-catenin or overexpression of the WNT inhibitor Dkk1 in the distal lung epithelium [90,111]. Remarkably, constitutively activating WNT signaling in embryonic lung endoderm using the Sftpc-Cre transgene results in a lineage switch with the presence of intestinal cells, including Paneth and goblet cells, in the pulmonary epithelium [87]. Hence, insight into metaplasia comes from knowledge of pathways regulating normal lineage commitment and differentiation in the developing lung.
CONCLUSION
Lung morphogenesis, like other developmental processes, depends on complex interactions that are tightly controlled both temporally and spatially. Extensive investigation over the past decade has uncovered select molecular processes that regulate different components of lung development. Table 1 recapitulates the principal factors involved and the genetic evidence that supports their role in lung morphogenesis and/or cell differentiation. Although the clinical relevance of these processes in human developmental anomalies and perinatal lung diseases remains to be fully elucidated, a better understanding of the molecular mechanisms of lung development will provide important insight into understanding congenital disorders of the respiratory tract, as well as controlling cell differentiation and regeneration for therapeutic purposes.
Pulmonary phenotypes in mutant mice
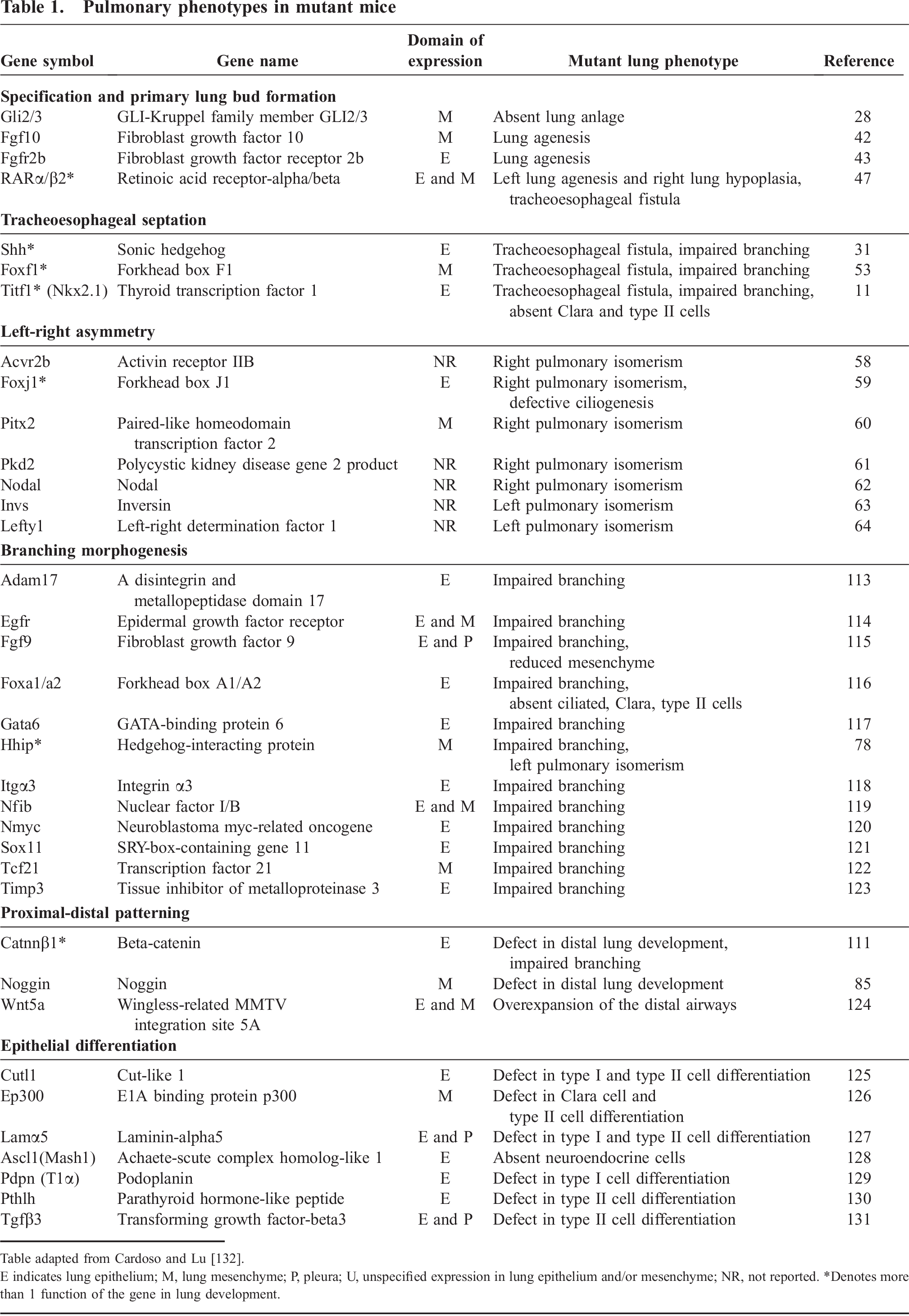
Table adapted from Cardoso and Lu [132].
E indicates lung epithelium; M, lung mesenchyme; P, pleura; U, unspecified expression in lung epithelium and/or mesenchyme; NR, not reported.
Denotes more than 1 function of the gene in lung development.