
Abstract
This review of pediatric pulmonary arterial hypertension provides a framework within which to view pulmonary hypertension in children. Classification schemes, including the latest recommendations from the World Health Organization, are discussed, and the histopathology of severe pulmonary hypertension is reviewed. New information is provided regarding idiopathic and familial forms of the disease. Specific childhood etiologies, including persistent pulmonary hypertension of the newborn and congenital heart disease, are reviewed. Additionally, we examine the role of collagen vascular diseases, portal hypertension, and viruses in the pathogenesis of severe pulmonary arterial hypertension.
Keywords
INTRODUCTION
Pulmonary arterial hypertension (PAH), whether idiopathic (IPAH) or secondary, is often a fatal disease in children and adults. It is clinically defined as a pulmonary arterial pressure >25 mm Hg at rest or >30 mm Hg after exercise. Clinical symptoms often are not apparent until the pulmonary artery pressure exceeds 60 mm Hg.
Romberg described the 1st case of pulmonary hypertension in 1891 and labeled it “pulmonary vascular sclerosis” [1]. In 1897, Eisenmenger provided the 1st link of pulmonary hypertension with congenital heart disease (CHD) in his detail of a 32-year-old patient with dyspnea and cyanosis at infancy and subsequent severe pulmonary hypertension and hemoptysis. Autopsy revealed a large ventricular septal defect and an overriding aorta [2], and the name “Eisenmenger syndrome” was later invoked to reflect pulmonary hypertension in patients with large left to right cardiac shunts [3].
In 1958, Heath and Edwards proposed a grading system for pulmonary hypertension, based on the severity of vascular changes observed in 67 children with CHD and 1 case of idiopathic pulmonary hypertension [4] (Table 1). Although this classification scheme was initially introduced to assess the potential reversibility of PAH associated with congenital heart defects, its utility has been debated, especially when applied to idiopathic and other forms of pulmonary hypertension [5]. Furthermore, this grading system implies a stepwise progression of disease from grade I to grade VI; yet it remains unclear whether such a progression occurs and, if so, whether it reflects severity or duration of pulmonary hypertension. In 1998, the World Health Organization (WHO) convened a panel of pulmonary hypertension experts and recommended that “the previous pathological classification of pulmonary vascular disease be abandoned. It has been found to be too restrictive and the classes and grades do not correlate with the clinical and hemodynamic findings in a consistent prognostic fashion outside of congenital heart disease.” [6]. Thus, although the Heath and Edwards grading system has utility in PAH associated with CHD, it has, in many ways, hampered investigations into the pathogenesis of other forms of PAH. To more accurately reflect current knowledge, the WHO Third World Conference on Pulmonary Hypertension in 2003 put forth a clinical-pathologic classification of pulmonary hypertension that emphasized recent rapid developments in pathogenetic mechanisms, including identification of familial forms of PAH (FPAH) (Table 2) [7].
Heath and Edwards grading scheme for hypertensive pulmonary vascular disease

From Heath and Edwards [4].
2003 Revised nomenclature and classification of pulmonary hypertension

Adapted from [7].
Although significant advances have been made in both basic and clinical research, especially with regard to FPAH (see below), pediatric PAH outside the setting of CHD remains a less studied, more enigmatic entity, stemming in part from the rarity of IPAH in children. Because of the few pediatric cases available for study, many of the adult pathogenetic mechanisms have been applied to childhood disease. However, as discussed below, the very young likely have a different pathogenesis of disease than do older children or adults.
SEVERE PAH
Histopathology of the vascular lesions in severe PAH
Pulmonary arterial hypertension results from structural alterations to the vasculature, affecting primarily arterioles and small to medium-sized pulmonary arteries. There are 3 main components of pulmonary arteries: the intima, the media, and the adventitia. Endothelial cells line the intima, smooth muscle cells constitute the media, and the adventitia is composed primarily of collagen and fibroblasts. All compartments of the vessel wall can be abnormal in severe PAH.
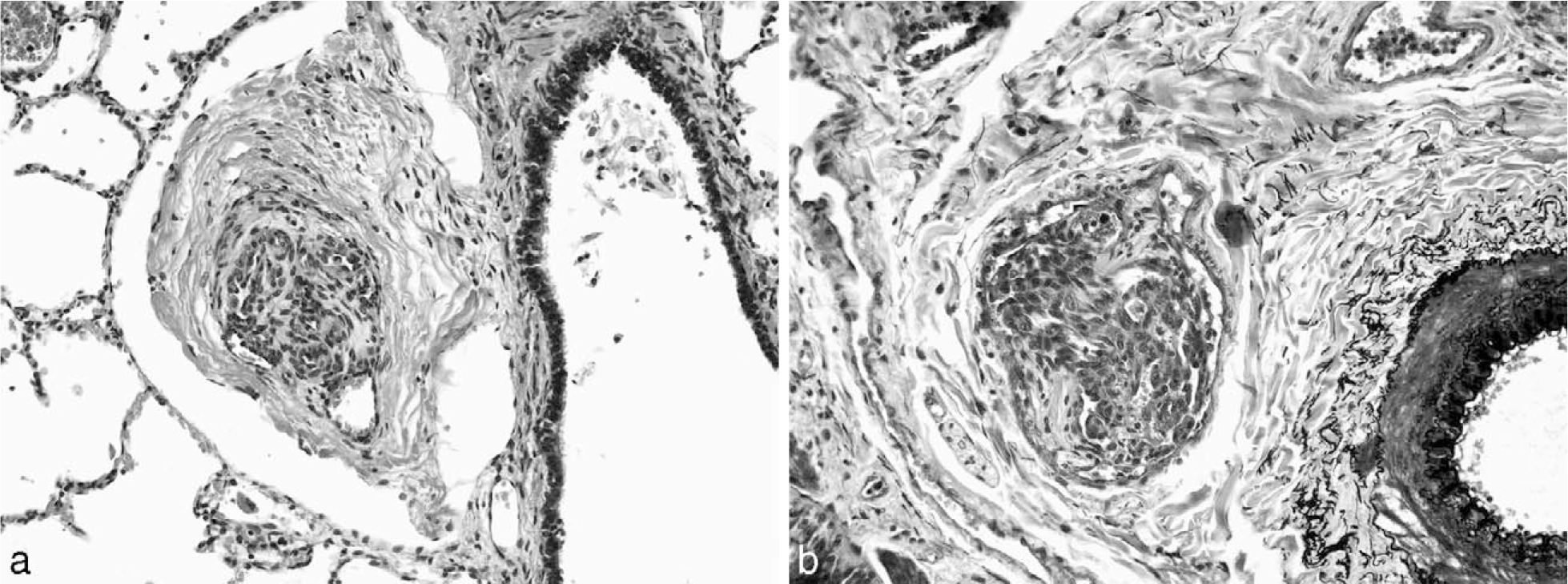
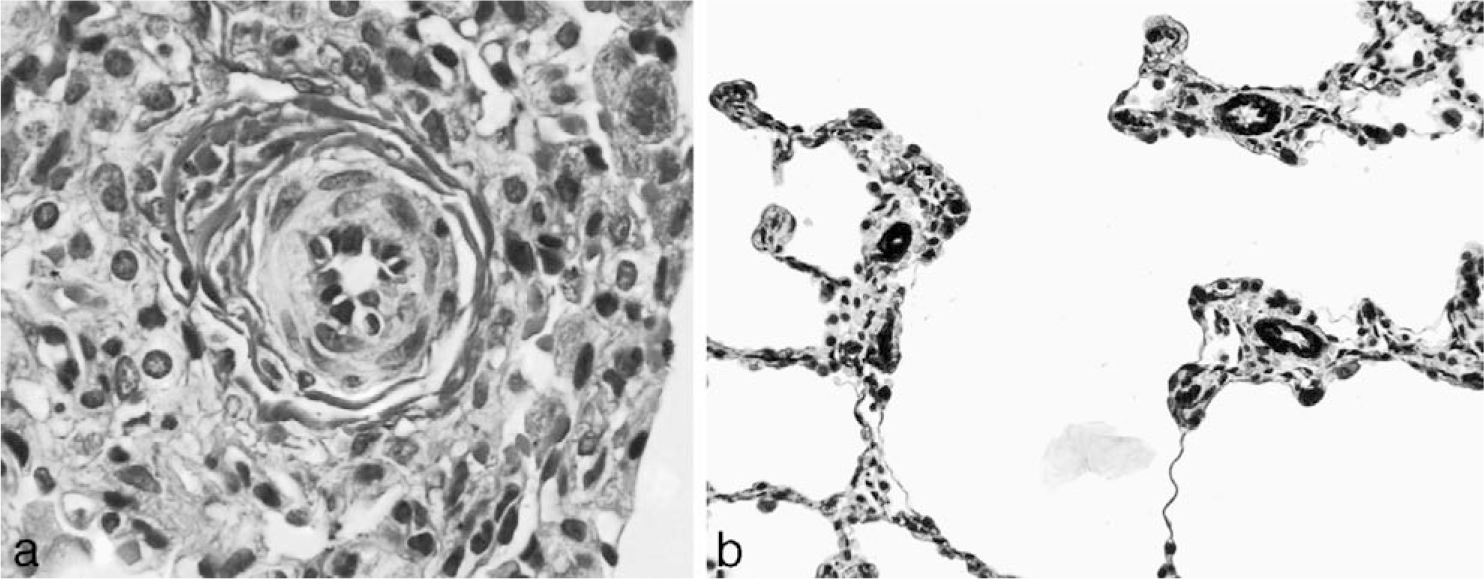
Vascular alterations of severe PAH are found in both IPAH and FPAH but also occur in association with CHD and other diverse diseases. Perhaps the most well-studied structural abnormality in severe PAH is the plexiform lesion (Fig. 1). Plexiform lesions are highly associated with severe clinical disease. The plexiform lesion is best characterized by an intraluminal proliferation of endothelial cells, which may be admixed with smooth muscle cells [8–10]. Often, secondary slit-like vascular lumina are found (in contrast to the more rigid, punched-out appearance of recanalized thrombotic arteries). Although the plexiform lesions may not be abundant, their particular location in branching points of small to medium-sized pulmonary arteries (Fig. 2) leads to profound disruption and obliteration of vascular lumens, causing severe pulmonary hypertension [9]. Based on 3-dimensional vascular reconstruction of plexiform lesions, the lesions are often preceded by short segments of concentric intimal fibrosis. Plexiform lesions can also occur as isolated lesions, without any evidence of intimal or medial vascular alterations [9]. Some authors hypothesize that the intraluminal proliferation of endothelial cells is analogous to a tumor-like proliferation of vascular cells [11]. The cells within the plexiform lesions of patients with IPAH have been extensively studied and found to show monoclonal cell growth [12], somatic endothelial cell mutations [13], loss of tumor suppressor genes [14], expression of antiapoptotic proteins [15], increased proliferation [16], and a whole host of other abnormalities associated with angioproliferation [17], including abnormal expression of vascular endothelial growth factor in the endothelial cells of the plexiform lesions. Most of this work has been performed in adult forms of severe PAH, and childhood forms of the disease will likely demonstrate other abnormalities. In fact, preliminary data suggest that the pathology and cellular composition of plexiform lesions in young children (<3 years of age) are different from those seen in older children or adults, in that young children have a more prominent smooth muscle component [18] (Fig. 3).
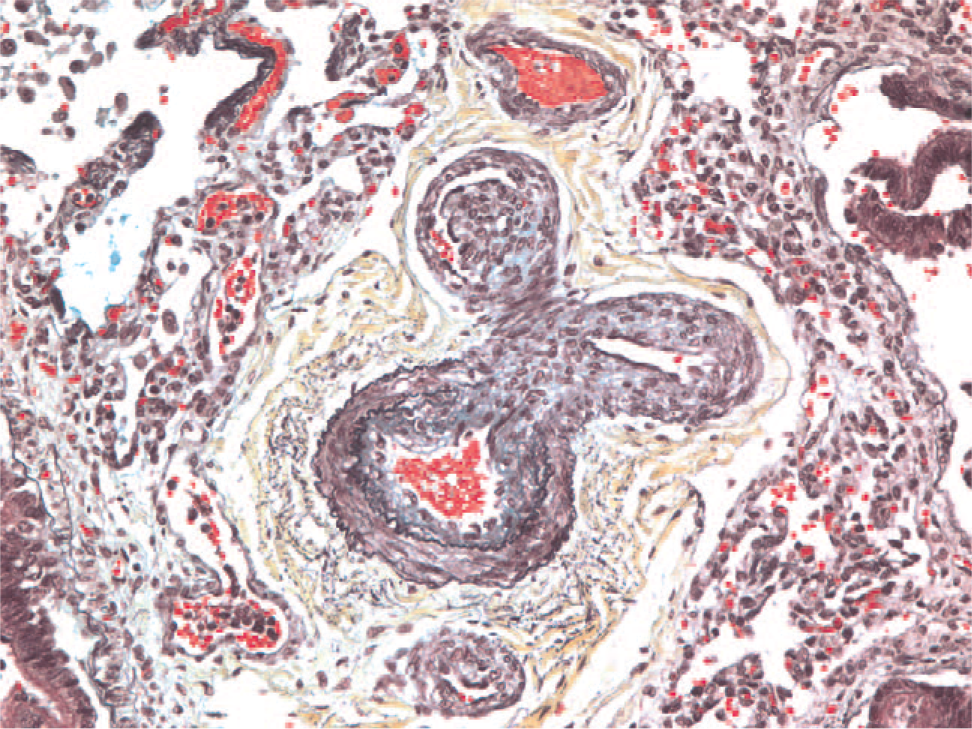
Pentachrome stain of a bifurcating pulmonary artery showing a plexiform lesion in the upper left branch. A color version of this figure is available online.
Although thickening of the smooth muscle layer (media) in the pulmonary arteries can be quite dramatic (Fig. 4), this does not necessarily correlate with the severity of clinical disease. Similarly, intimal hyperplasia can be a nonspecific change and may be seen in any lung disease that leads to hypoxia. Patients with interstitial lung disease, for example, may have secondary vascular changes that may not be associated with clinically apparent or significant pulmonary hypertension.
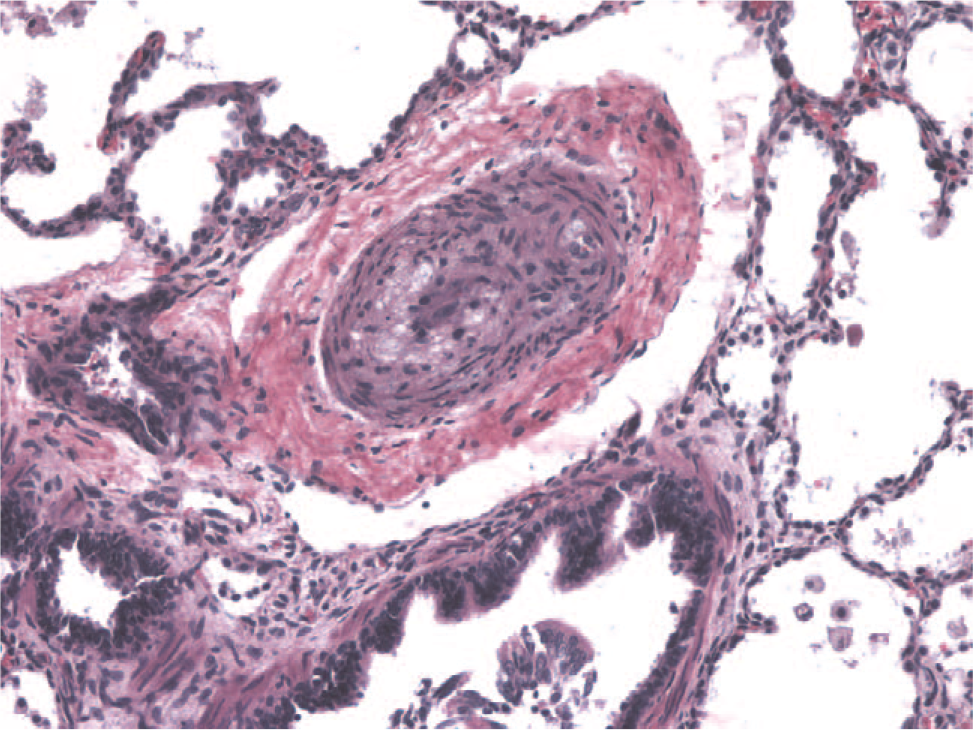
Hematoxylin and eosin–stained section of the pulmonary artery from a 1-month-old child with hypoplastic left heart. The smooth muscle is hypertrophied, and the lumen is nearly occluded. A color version of this figure is available online.
In contrast to larger vessels, extension of smooth muscle into arterioles (<50 μm) can be associated with striking clinical pulmonary hypertension, including in young children with cardiac anomalies (Fig. 5). Unlike the patchy nature of the plexiform lesions in severe PAH, muscularization of arterioles is often a more diffuse process but may be a subtle histologic change.
Idiopathic (primary) pulmonary arterial hypertension
Cases of IPAH are common in young females (mean age 36.4 years), although the disease can occur at any age and in either sex [19]. The incidence of disease is estimated to be 1 to 2 cases per million, but in children the incidence is even lower [19]. Most work in IPAH has been performed in adults, and the disease is not well understood in young children. Although initially thought to follow the progression of lesions in the Heath and Edwards classification scheme (Table 1), it is now known that plexiform lesions can appear as the sole histologic manifestation of disease and may not be related to any other vascular abnormality, including smooth muscle hypertrophy.
Because the natural history of the plexiform lesion in disease is not known, the pathogenesis of the plexiform lesion is also not known. Because it is difficult to biopsy patients with severe PAH, most of the work to date has been in experimental animal models and in autopsy material from patients who died of their disease. Animal models have been studied for years, but very few mimic the vascular lesions found in humans [20,21]. Most animal models of PAH show arterial smooth muscle thickening as a cause of pulmonary hypertension, unlike much of the human pulmonary vascular disease, which shows a significant endothelial cell component with plexiform lesion formation. Therapeutic strategies in patients with severe PAH have reflected this bias toward smooth muscle hypertrophy as a drug target. Although many of the drugs (such as prostacyclin) that have increased survival in patients were initially thought to act mainly as vasodilators, their ability to dilate pulmonary arteries has proved to be modest. It is now known that prostacylin likely has a pleiotropic effect, modifying both endothelial function and smooth muscle proliferation. Pulmonary arterial hypertension results from an imbalance of growth suppressors and promoters, and prostacylin partially restores that imbalance. Now, new therapies (such as endothelin-1 antagonists) are available to treat the endothelial cell growth component of the disease.
The pathology of IPAH is similar to that seen in associated forms of the disease. There are scattered plexiform lesions within small to medium-sized pulmonary arteries. The medial contribution ranges from minimal to marked smooth muscle hypertrophy. Young children will often have marked muscularization of arterioles. In patients who are not anticoagulated, there may be scattered thrombi in vessels. Recanalization of vascular lumens may mimic plexiform lesions, but immunohistochemical stains for endothelial cells and smooth muscle cells can help highlight the increased endothelial cell contribution in IPAH (Fig. 6).
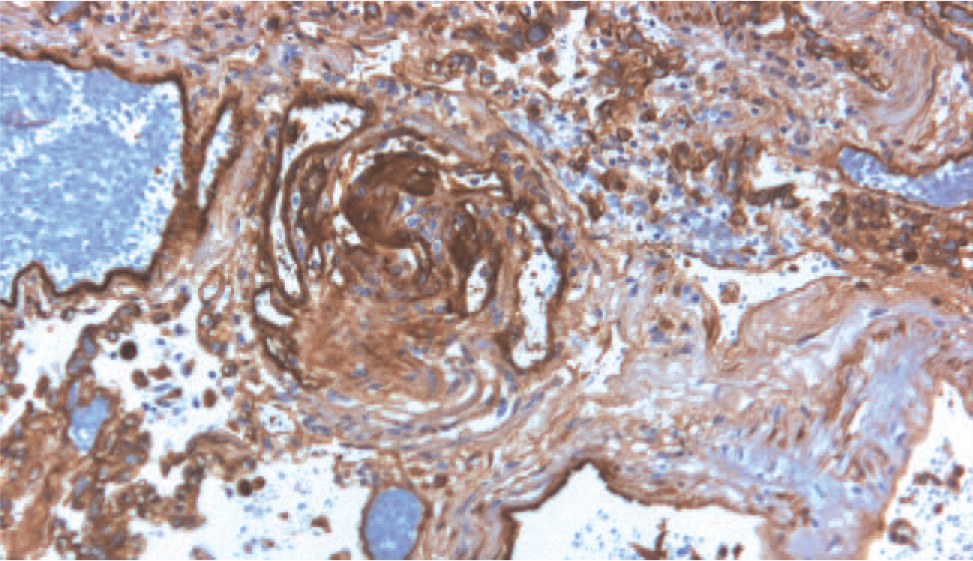
Immunohistochemical stain for the endothelial cell marker, CD31, which decorates the prominent endothelial cell component of a plexiform lesion in an adult patient with idiopathic pulmonary arterial hypertension. A color version of this figure is available online.
Our understanding of the etiologic mechanisms in IPAH remains incomplete. Centralized tissue collection centers have been established to facilitate study of the disease. Much of the current research is focused on better understanding the genetic contributions to the disease, which are discussed below.
Familial PAH
Familial pulmonary arterial hypertension accounts for less than 6% of primary (idiopathic) cases [19]. The disease is autosomal dominant with incomplete penetrance and, in 50% of the cases, linked to germ-line, exonic mutations in bone morphogenetic protein receptor II (BMPR2), found on chromosome 2q31–32 [22–26]. Bone morphogenetic proteins are part of the transforming growth factor β (TGF-β) superfamily of cytokines. Mutations in BMPR2 confer a 15% to 20% chance of developing PAH in a carrier's lifetime. The familial form of the disease appears at a younger age in each successive generation (genetic anticipation) and is more common in females, despite the autosomal dominant pattern of inheritance.
Not only have BMPR2 mutations been identified in FPAH but they have also been identified in a subset (26%) of IPAH patients [27,28]. Additionally, in 18 children younger than 6 years of age who had IPAH (n = 16) or PAH associated with CHD (n = 2), 4 of the patients (all with IPAH) demonstrated germline mutations in genes encoding receptor members of the TGF-β cell-signaling pathway [29]. Mutations in an activin receptor–like kinase, another member of the TGF-β superfamily, have been found in patients with pulmonary hypertension associated with hereditary hemorrhagic telangiectasia [30]. Other genes certainly remain to be identified, and because only 15% to 20% of family members with the BMPR2 mutation develop disease, other mechanisms must be involved in the formation of clinical disease.
The histopathology of FPAH is indistinguishable from IPAH in that plexiform lesions and muscularization of arterioles are common to both forms of the disease. Medial hypertrophy of pulmonary arteries can also be prominent in both FPAH and IPAH.
WHAT ARE SPECIFIC CHILDHOOD ETIOLOGIES ASSOCIATED WITH THE DEVELOPMENT OF SEVERE PAH?
Persistent pulmonary hypertension of the newborn
Persistent pulmonary hypertension of the newborn (PPHN) is multifactorial in origin and may be idiopathic (rare) or associated with congenital heart disease or hypoxic lung disease (acquired or congenital). The relationship of PPHN to IPAH is unclear.
Alveolar capillary dysplasia with misalignment of pulmonary veins (ACD-MPV) has been increasingly recognized as a cause of PPHN [31,32]. Most patients with ACD-MPV present in the 1st few days of life with cyanosis, respiratory distress, and PPH. There is no sex predilection, and the disease is invariably fatal. One study showed that 6 of 13 newborn infants with PPH not associated with any known cause had ACD-MPV at autopsy [33]. Two of the 6 infants had surgical lung biopsies, which showed histologic changes of ACD-MPV, suggesting that ACD-MPV can be diagnosed during life.
Histologically, PPHN is characterized by persistence of a fetal arterial wall structure. Normally, immediately after birth, the pulmonary arterial wall becomes thin and the lumen is open. With PPHN, there is failure to appropriately remodel the vessel wall, and the media hypertrophies instead of thins. Without treatment (and sometimes with), the medial hypertrophy leads to vascular obstruction.
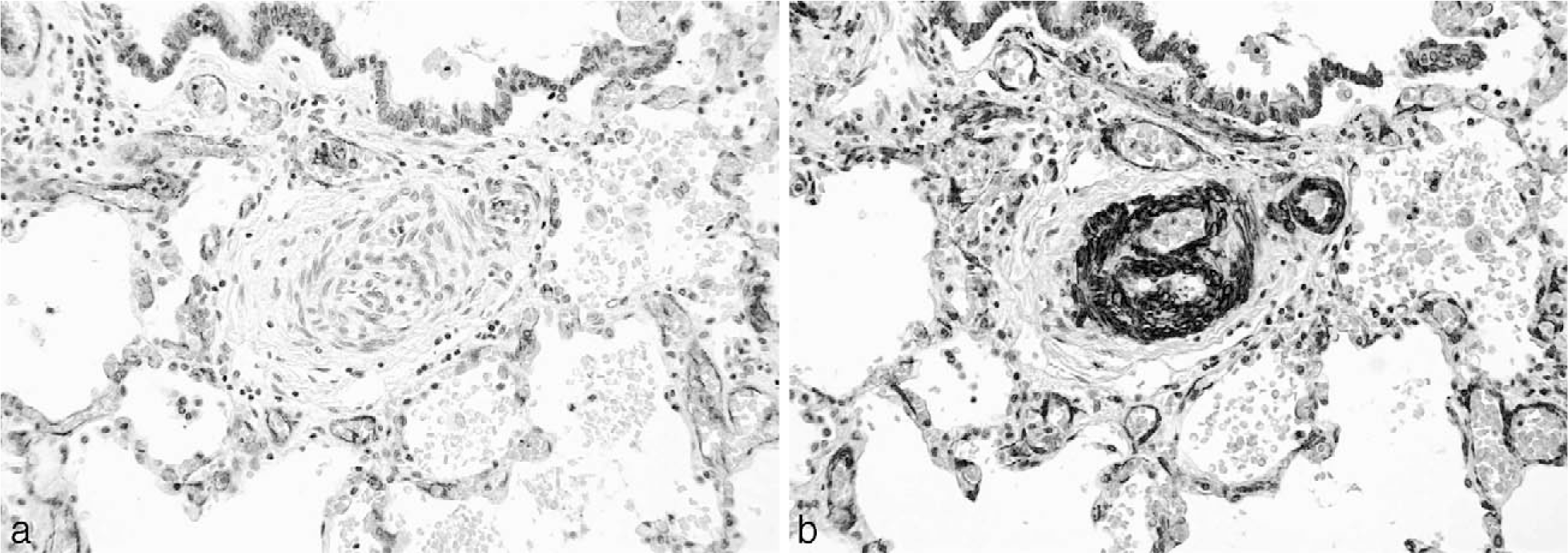
In ACD-MPV, the small pulmonary arteries adjacent to small airways and within the lobular parenchyma show prominent muscularization (Fig. 7). The accompanying anomalous, malpositioned veins and venules are dilated and often congested (Fig. 8). Arterioles are muscularized. There are decreased numbers of alveolar capillaries and interlobular septal veins. Pulmonary lymphangiectasia accompanies one third of the cases [34].
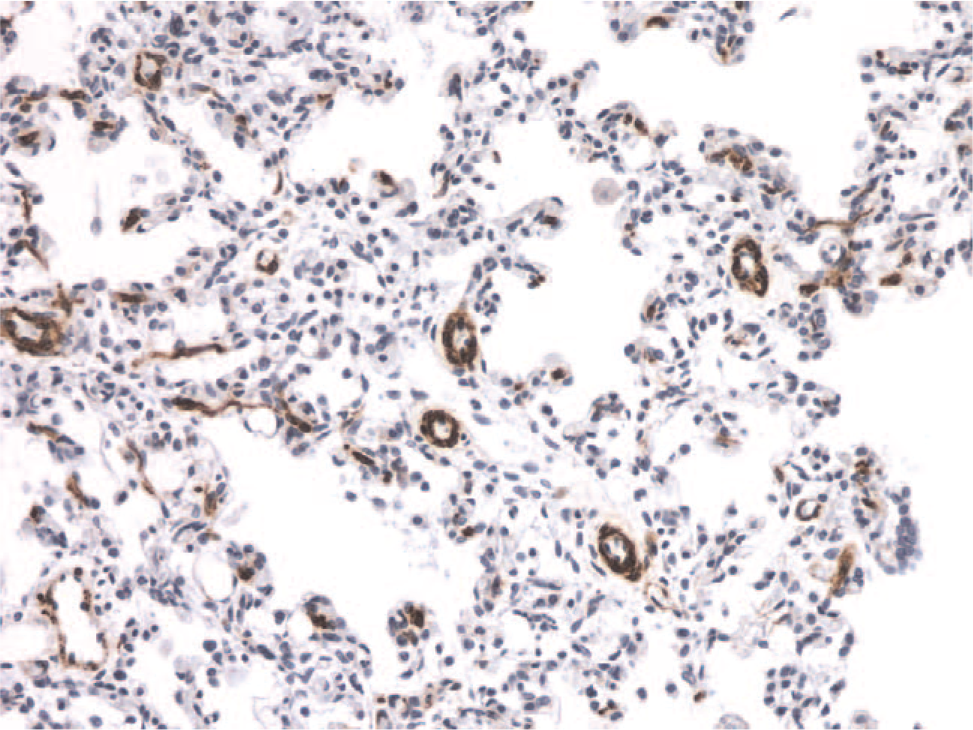
Example of muscularization of the distal pulmonary arterioles in a patient with persistent pulmonary hypertension of the newborn. The muscle-specific antibody immunostain highlights the abnormality of the smooth muscle in the vessel walls. A color version of this figure is available online.
Pentachrome stain demonstrating the malpositioned vein that is situated between the pulmonary artery (bottom) and bronchiole (top). A color version of this figure is available online.
Congenital heart disease
Unlike PPHN, which is evident early on, the pulmonary vascular disease associated with CHD takes time to develop. Neonatal disease is unusual. The rate of development of pulmonary vascular disease is dependent on the type of CHD and the underlying genetic predisposition of the patient. Not everyone with CHD develops PAH. Approximately 50% of infants with a large, nonrestrictive ventricular septal defect or pulmonary ductus arteriosus develop pulmonary hypertension by early childhood [35,36]. Forty percent of patients with ventricular septal defect or pulmonary ductus arteriosus and transposition of great vessels develop PAH within the 1st year of life [36]. Other CHDs that lead to progressive PAH include large secundum atrial septal defects, persistent truncus arteriosus, and common atrioventricular canal.
How does increased blood flow through the pulmonary circulation lead to PAH? Untreated congenital cardiac defects with intracardiac communications eventually lead to systemic or suprasystemic levels of pulmonary flow with subsequent development of pulmonary vascular disease (Eisenmenger syndrome). Although the size of the vessels affected in PAH suggests that only laminar flow occurs, local turbulence and mechanical stress likely lead to alterations in expression of specific genes in the endothelial cells lining the vessels. The specific localization of plexiform lesions at the bifurcation points of pulmonary arteries also suggests a relationship to increased shear forces.
The altered shear forces acting on the endothelial cells can lead to upregulation of growth factors, such as vascular endothelial growth factor. Indeed, vascular endothelial growth factor is one of the factors upregulated in in vitro endothelial cells exposed to increased shear stress [37].
Why do some children develop irreversible pulmonary vascular disease, and others show complete reversal after repair? A recent study by Lévy et al showed that irreversible PAH in children with high-risk CHD was strongly associated with impaired endothelial cell apoptosis and antiapoptotic signaling from perivascular inflammatory cells [38]. Patients with both reversible and irreversible pulmonary hypertension expressed proapoptotic markers in their endothelial cells; however, only patients with irreversible pulmonary hypertension expressed the antiapoptotic protein Bcl-2. The authors hypothesize that the apoptosisresistant endothelial cells lead to more intimal proliferation and occlusion of distal arteries.
Nowhere is the Heath and Edwards grading scheme more appropriately applied than in patients with CHD as a cause of PAH. However, because pulmonary angiography and measurement of pulmonary vascular hemodynamics have become commonplace, the role of lung biopsy in patients with CHD is limited.
WHAT ARE OTHER POTENTIAL ETIOLOGIES ASSOCIATED WITH THE DEVELOPMENT OF SEVERE PAH?
Collagen vascular diseases
Children, like adults, develop all forms of collagen vascular diseases. The most common secondary form of PAH is due to collagen vascular disease. Pulmonary arterial hypertension develops most often in patients with the limited form of systemic sclerosis or the calcinosis cutis, Raynaud phenomenon, esophageal dysmotily, sclerodactyly, telangiectasia (CREST) syndrome. Up to 50% of patients with the CREST syndrome develop PAH [39,40]. A large study of patients with systemic sclerosis (without pulmonary fibrosis) demonstrated that 12% (89/722) had PAH by right heart catheter [41]. Other studies show a range of prevalence from 2.3% to 35% [39,42,43]. Similar incidences have been found in patients with mixed connective tissue diseases [44,45]. In patients with rheumatoid arthritis, up to 21% have been reported to develop PAH [46,47]. Although less commonly associated with systemic lupus erythematosus, the prevalence reports vary from 0.5% to 14% [48–51].
The mechanisms that lead to the development of PAH in collagen vascular diseases are not well understood. Microvascular injury likely leads to endothelial cell dysfunction and proliferation. Immunologic abnormalities may also contribute to the development of isolated pulmonary hypertension in patients with collagen vascular diseases. A number of studies have reported a prevalence of anticardiolipin antibodies in patients with collagen vascular diseases and pulmonary hypertension [49,52–55].
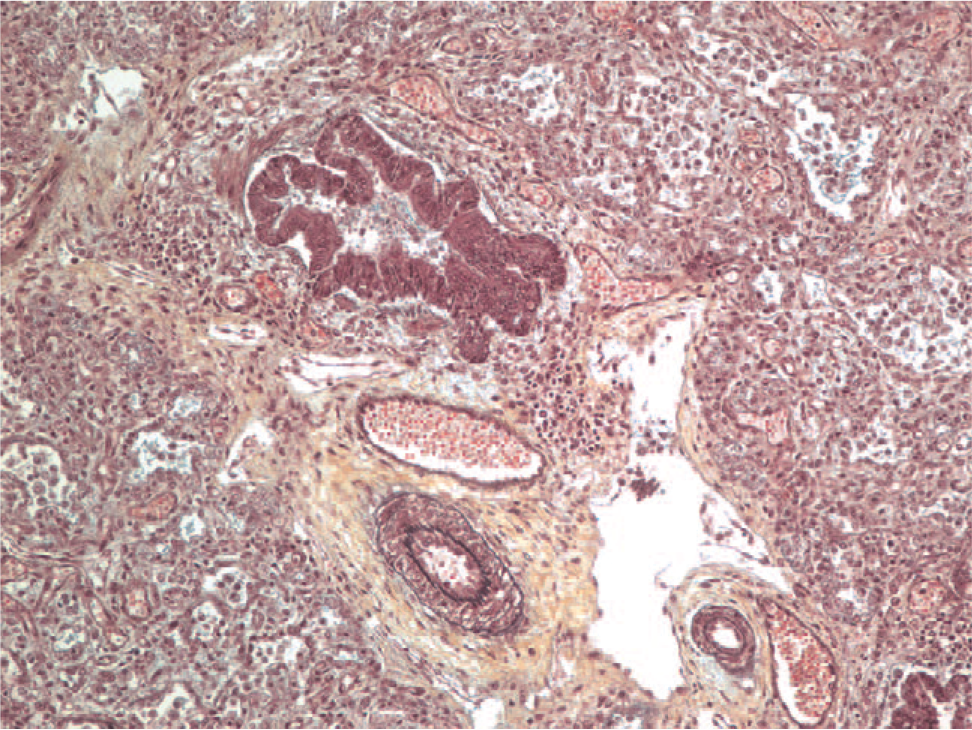
The pathology of pulmonary hypertension related to a collagen vascular disease can often be discerned by the presence of interstitial or pleural abnormalities associated with the collagen vascular disease. For example, lymphoid hyperplasia is seen with rheumatoid arthritis. Scleroderma can show a nonspecific interstitial pneumonia pattern of fibrosis. Often the vascular changes include a more pronounced, collagenized adventitia, as well as more prominent small-vessel disease (Fig. 9). Plexiform lesions can be sparse and difficult to find. Although intimal hyperplasia is usually a relatively nonspecific finding, a rare form characterized by concentric laminar intimal hyperplasia/fibrosis (“onionskinning”) is often a clue to an underlying collagen vascular disease [56].
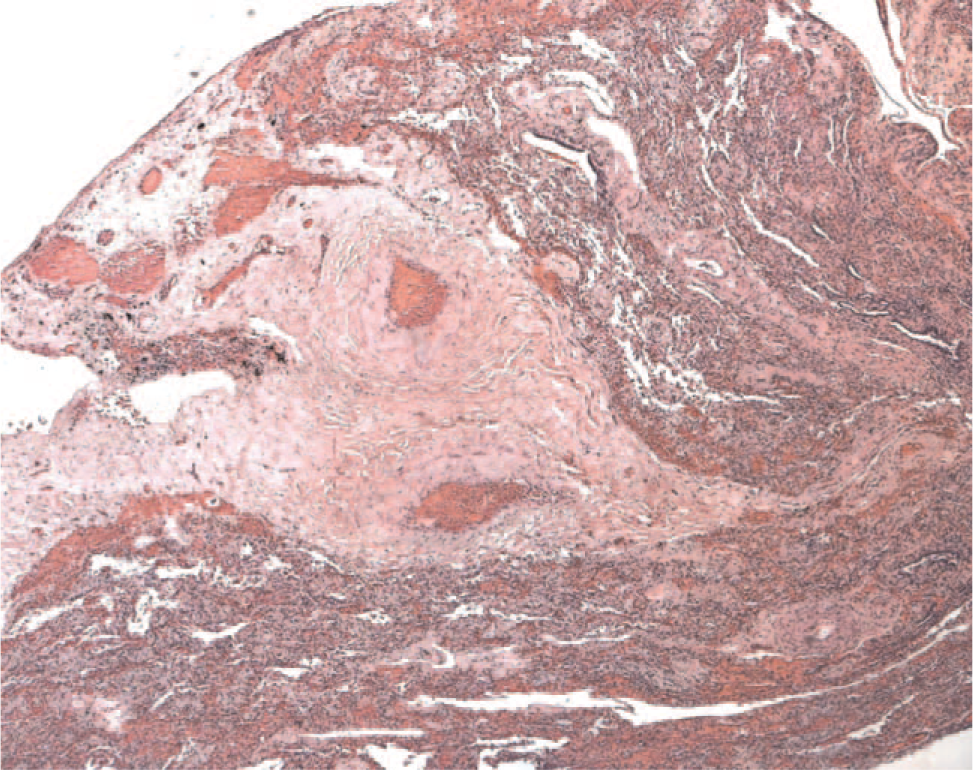
Hematoxylin and eosin–stained section from an adult patient with scleroderma demonstrating the marked adventitial collagen. The vessels appear to be floating in a sea of hyalinized collagen. The pulmonary artery lumens are patent and filled with red blood cells. A color version of this figure is available online.
Portal hypertension
The incidence of PAH in portal hypertension (known as portopulmonary hypertension) is between 2% and 5% [57]. In those patients undergoing liver transplant, the prevalence has been shown to be 8.5% [58]. Although the pathogenesis of the arterial hypertension is not known, postulated mechanisms include circulating “toxic” vasoactive factors leading to endothelial cell injury and proliferation [57,59,60], increased blood flow causing vascular shear stress [61], and increased levels of endothelin-1, which not only is a potent vasoconstrictor but also is produced by both liver and lung [62–64]. The development of portopulmonary hypertension also appears to be independent of the cause of portal hypertension [65]. The risk of developing PAH is associated with the duration, not degree, of portal hypertension.
Portopulmonary hypertension affects children as well as adults. A recent report illustrates the necessity to screen for this disease in children prior to liver transplant, because 4 of the 7 children reported in the series died [66].
The pathology of pulmonary vascular disease in portal hypertension is indistinguishable from that found in IPAH and FPAH. Plexiform lesions, intimal hyperplasia, medial hypertrophy, and muscularization of arterioles can all be seen in patients with severe PAH.
Viruses
The incidence of PAH in the human immunodeficiency virus (HIV) population is about 0.5%, which is a 2000-fold increase in risk compared with the general population [67]. A recent report finds a high prevalence of PAH in children infected with HIV [68]. The virus itself has never been found in the lungs of patients with PAH, suggesting that HIV acts indirectly on the endothelial cells by producing growth factors and cytokines [69].
Recent reports examined the potential role of human herpesvirus-8 (Kaposi's sarcoma virus), a gamma herpes virus, in patients with PAH. Two patients with Castleman's disease (which is caused by HHV-8 infection in 50% of the cases) were diagnosed with PAH [70]. In 16 patients with IPAH, 10 were positive for HHV-8, whereas only 1 of 14 patients with secondary PAH was positive for the virus [71]. However, subsequent studies at other centers have not confirmed HHV-8 infection in patients with PAH [72–75], including pediatric IPAH [76]. Human herpesvirus-8 infection varies widely from region to region, however, and its association with PAH may be related to regional, genetic, and/or environmental differences in the virus.
Although there is a clear-cut association between HIV infection and development of PAH, other viral etiologies remain to be confirmed.
CONCLUSIONS
Although PAH is a heterogeneous disease, the pathology in most cases is similar. There are minor differences, however, in that young children show a more prominent smooth muscle component and patients with collagen vascular diseases (such as scleroderma) often show marked adventitial changes. Older children are similar to adults in pathology of the vascular lesions. Additionally, older children and teenagers have similar pathogeneses to that of adults, whereas younger children likely have different etiologies and pathogenetic mechanisms.
Classification schemes for PAH have changed throughout the years and continue to evolve with the discovery of molecular mechanisms. The genetic clustering of patients with severe PAH led to identification of a region in the short arm of chromosome 2 and subsequent discovery of the BMPR2 mutation in a majority of the patients with FPAH and a minority of the patients with IPAH. These discoveries provide the foundation for future work in the genetic contributions to the disease.
Although genetic susceptibility is undoubtedly a key factor in the eventual development of disease, other factors, including collagen vascular diseases/autoimmunity, portal hypertension, viruses, increased blood flow, and persistence of fetal vasculature, can also lead to severe PAH. The relative contributions of genetic susceptibility and underlying/superimposed disease remain to be elucidated.
Survival in older children and adults has increased considerably based on current treatment regimens. However, despite the advance of medical management in severe PAH, there is no cure, and lung transplantation is often the only viable treatment. Clearly, if the mechanisms leading to the development of the vascular occlusive lesions can be identified, then new treatment strategies targeted at the microvascular lesions will be able to provide increased survival and a potential cure. Pulmonary arterial hypertension research is rapidly evolving, and it is likely that the next decade will provide rapid advances in gene discovery and treatment.