
Abstract
We present the 1st case report of an additional enteric smooth muscle layer in a patient with Mowat-Wilson syndrome and Hirschsprung disease. After resection of the aganglionic colon at the age of 5 months, our patient initially suffered from intermittent constipation, and subsequently by the age of 5 years, he developed ongoing diarrhea requiring medical treatment for more than a decade. Although the exact mechanism of abnormal gut motility in this case is unknown, we postulate that the supernumerary muscle and its associated neural plexus may be responsible for the patient's unusual late complication in treated Hirschsprung disease.
INTRODUCTION
Mowat-Wilson syndrome (MWS) is a multiple congenital anomalies/mental retardation syndrome caused by deletions or mutations in the ZEB2 (zinc finger E-box-binding homeobox 2) gene located on chromosome 2q [1]. The syndrome was 1st described in 1998 by Mowat and colleagues [2]. It is characterized by distinct facial features (large, medially flaring eyebrows; deep set, large eyes; hypertelorism; large, uplifted ear lobes with central depression; broad nasal bridge; prominent rounded nasal tip; open mouth; prominent columella and chin), developmental delay/mental retardation, microcephaly, and seizures [1,2]. In the initial series described by Mowat and colleagues, 5 of 6 had Hirschsprung disease [2]. In 2007, Garavelli and colleagues showed that Hirschsprung disease affected 57% of patients with MWS [1].
We report a boy with MWS including Hirschsprung disease in whom an additional layer of smooth muscle within the submucosa in the colon was found. Following Duhamel rectosigmoidectomy, our patient requires ongoing medical treatment for persistent diarrhea.
CASE REPORT
Clinical History
A 4-week-old male infant presented with a 2-day history of constipation, abdominal distension, and vomiting. Before presentation, he had an uneventful neonatal period. He passed meconium within 24 hours of a term delivery. He had daily bowel movements in the 1st week of life followed by passage of loose stools twice daily until presentation. A rectal suction biopsy confirmed Hirschsprung disease, and a sigmoid colostomy was formed. Large bowel removed at that time revealed ganglion cells and an extra layer of smooth muscle in the submucosa. At 5 months of age, Duhamel pullthrough at the level of sigmoid colon was performed. The resected rectosigmoid colon again showed an additional layer of smooth muscle in a segmental distribution.
For approximately 3 years after the Duhamel rectosigmodiectomy, he suffered from constipation and required a rectal washout at the age of 3 years. Subsequently, he developed diarrhea by the age of 5 years, which has persisted since. Upper gastrointestinal and rectal endoscopic biopsies at 10 years of age showed no significant abnormalities or cause for his symptom. Follow up at 16 years of age revealed that he passed about 4 loose stools a day despite loperamide.
In addition to Hirschsprung disease, he was noted to have global developmental delay and microcephaly at the age of 1 year. He developed seizures at 2 years of age. When he was 10, he had bilateral orchidopexies. A diagnosis of MWS was made at the age of 15 years when genetic testing confirmed a ZEB2 mutation. However, the precise details of the mutation/deletion are not available.
Histopathology
The rectal suction biopsy at the age of 4 weeks showed the typical features of Hirschsprung disease comprising increased number of thick nerves in the submucosa with absence of ganglion cells. The mucosa was normal.
Sigmoid colon resected at the time of stoma formation (age 4 weeks) showed the presence of both ganglion cells and prominent nerves indicative of transitional zone. In addition, an extra layer of smooth muscle of variable thickness (up to 0.1 mm thick) and fiber orientation was noted in the submucosa separated by fibrous connective tissue from the circular muscularis propria and muscularis mucosae (Figs. 1,2). Neuronal plexus containing ganglion cells were scattered within this extra coat of muscle (Fig. 3). No nerve trunks were identified within the additional layer. A normal myenteric plexus was present between the circular and longitudinal muscle.
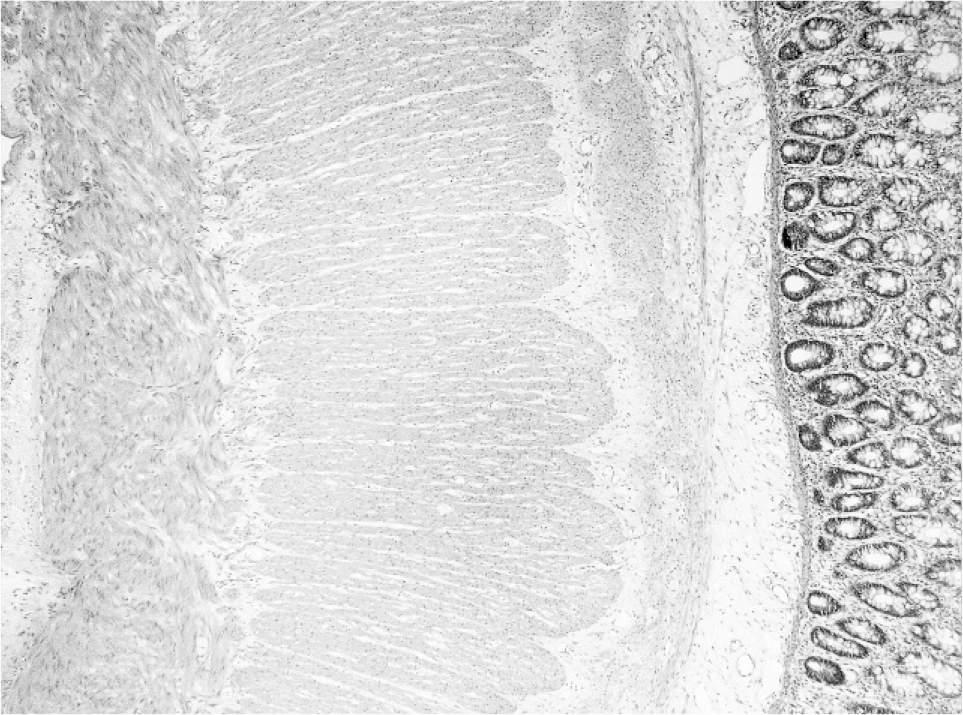
A full-thickness section of colon stained with hematoxylin and eosin shows an additional muscle layer in the submucosa.
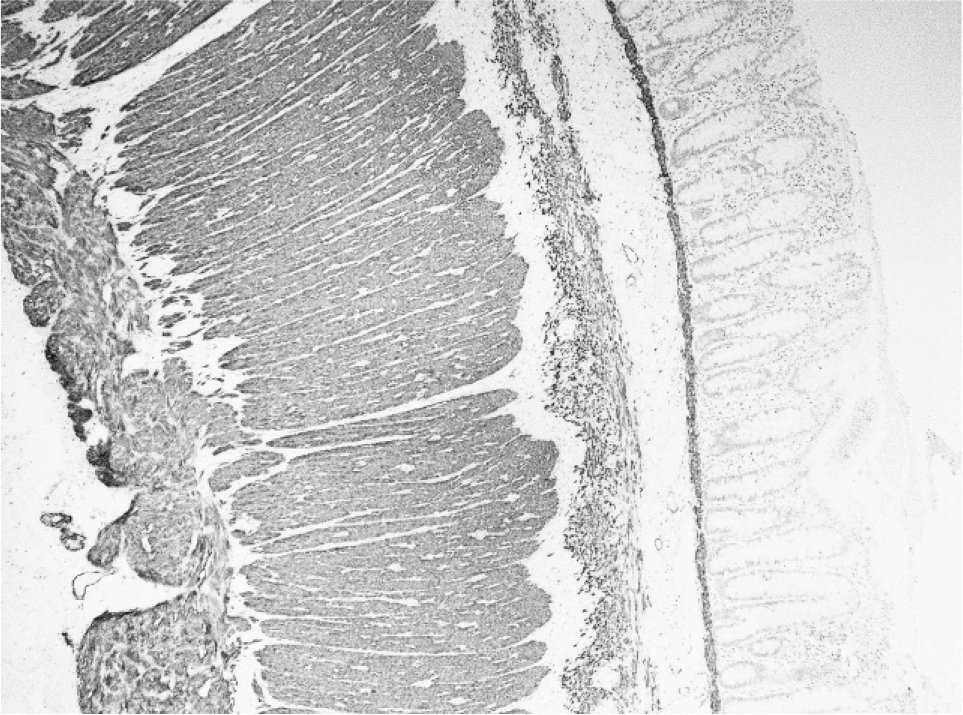
Desmin immunohistochemistry highlights the additional muscle layer in the submucosa.
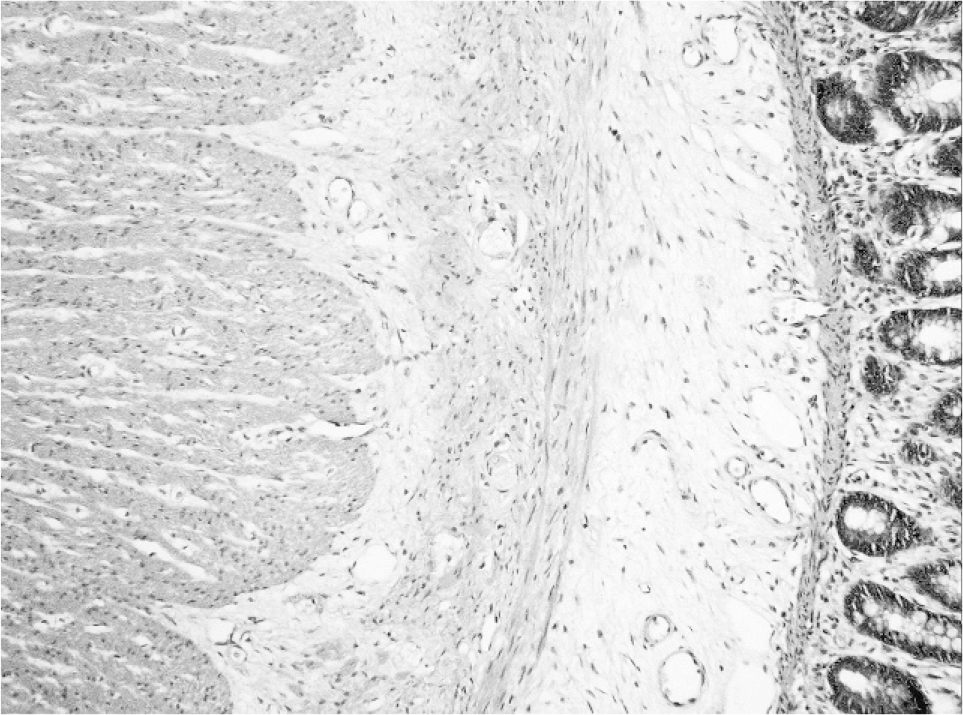
Scattered ganglion cells are present within the additional muscle coat.
Rectosigmoid colon (6 cm in length) and colostomy resected at Duhamel procedure at the age of 5 months revealed the presence of the additional smooth muscle layer in the aganglionic segment (distal, 2 cm), the proximal resection margin (ganglionic, 0.5 cm in length), and the stoma. Thus, the supernumerary coat was incompletely resected.
The presence of a muscle abnormality in the endocopic upper gastrointestinal and the rectal biopsies cannot be assessed, because these are made up of mucosa only.
DISCUSSION
Mowat-Wilson syndrome is a relatively recently recognized multiple congenital anomaly/mental retardation syndrome caused by heterozygous deletions or mutations of the ZEB2 (zinc finger E-box–binding homeobox 2 gene, previously called ZFHX1B) gene on chromosome 2q, which encodes for SIP1 (Smad-interacting protein 1, SMADIP1) [1]. The prevalence of MWS is currently unknown. Approximately 171 patients with ZEB2 mutations, deletions, or cytogenetic abnormalities have been reported [1]. More than 100 deletions/mutations have been described to date in patients with a typical phenotype of MWS [1]. It was 1st described in 1998 in 6 children with distinctive facial dysmorphic features, mental retardation, seizures, microcephaly, and short stature. Five of these children were shown to have Hirschsprung disease, and the other child had chronic constipation [2]. Hirschsprung disease was reported in 57% of published cases of MWS [1]. Both short- and long-segment diseases were reported in both sexes. It was noted that patients with ZEB2 deletion, but not those with mutations, tend to develop more extensive aganglionosis [3]. Another gastrointestinal anomaly noted in MWS is pyloric stenosis [1]. None of the reports have mentioned an additional muscle layer when referencing the histology of patients with this syndrome.
The presence of an additional layer of smooth muscle in the intestine is rare. To the best of our knowledge, there have been 9 cases reported [4–8]. Four cases showed the extra layer in the luminal side of the muscularis propria [5,6,8]. The extra muscle in 2 cases was external to the longitudinal muscle [4,8], and 3 cases showed division of the circular muscle into 2 layers by neural plexus [5].
Smith and colleagues described 5 patients with an additional intestinal muscle coat and classified them into 2 histologic groups: 3 related male patients in type 3 “diffuse familial abnormal muscle layering” and 2 unrelated female patients into type 4 “segmental additional muscle coat” [5]. The 2 female patients demonstrated similar histopathologic changes to our case, showing an additional muscle layer on the inside of the circular muscle associated with an additional neural plexus between the extra layer and the circular muscle in the colonic wall. Similar morphologic changes were reported by Kapur and colleagues, Hart and colleagues, and Goulet colleagues [6–8].
Kapur and colleagues reported 2 patients presenting with intestinal pseudo-obstruction and architectural malformation of their intestinal muscularis propria [8]. One of their patients, patient 1, presented with constipation and had similar morphologic appearances to those seen in our patient.
Hart and colleagues reported a 23-year-old man with lifelong constipation and fecal soiling who had an additional layer of smooth muscle containing neurons on the luminal side of the muscularis propria in the resected megacolon. The patient's symptoms improved significantly after a colectomy [6].
Goulet described a “3rd muscle layer” in a child with pseudo-obstruction, but no further clinical or histologic details were provided [7].
Ours is the 1st case of additional intestinal smooth muscle found in a patient with MWS and Hirschsprung disease. Our patient is also unusual in that he suffers from diarrhea, unlike other patients, who are reported to have symptoms of pseudo-obstruction.
Bonnard and colleagues evaluated the clinical course and outcome of 5 patients with Hirschsprung disease associated with MWS [9]. All 5 patients and our patient were full-term babies. Interestingly, 4 of 5 patients (80%) passed meconium within the 1st 24 hours, as did our patient, compared with less than 10% of children with Hirschsprung disease passing meconium during that time [10]. Two of the 5 patients had rectosigmoid involvement, and 3 had near total/total colonic disease. Two patients with total colonic disease had several episodes of enterocolitis and diarrhea during follow up [9]. Our patient, on the other hand, has been suffering from persistent diarrhea for more than a decade after definitive treatment for aganglionosis.
Diarrhea, not associated with enterocolitis after resection of aganglionic colon, is an unusual feature. Well-recognized, late complications in children with treated Hirschsprung disease are ongoing obstructive symptoms, incontinence, and enterocolitis [10]. The finding of supernumerary enteric muscle coat with its associated neural plexus in our patient leads us to speculate that the abnormality may be contributory to hypermotility manifesting as diarrhea.
Acquired muscularization of the submucosa can be seen in Crohn disease and in ischaemic and radiation enteritis [11]. Two postulated mechanisms are (1) direct muscularization of fibrotic submucosa and (2) proliferation and fusion of reparative muscularis mucosae and muscularis propria in the vicinity of ulceration [11]. The injury-related submucosal smooth muscle hyperplasia is thought to arise from the muscularis mucosae and extend into the submucosa and is usually associated with fibrous tissue. The extra muscle coat in our case was composed, at least focally, of discrete, large bundles of smooth muscle in the deep submucosa with no evidence of fibrosis, inflammation, or other previous damage. The additional muscle has been considered to be a congenital abnormality due to a primary defect in morphogenesis of the enteric smooth muscle and/or enteric nervous system. EB2 functions as a DNA-binding transcriptional regulator that interacts with activated SMADs, the intracellular proteins that act as transducers of transforming growth factor beta signaling [12]. Transforming growth factor beta superfamily family members, which include activin and bone morphogenetic proteins, exert a variety of effects, including cell proliferation, differentiation, migration, adhesion, and apoptosis. ZEB2 mRNA is expressed at varying levels in multiple tissues, the highest intensity being detected in the central and peripheral nervous system [13]. Thus, SIP1 is a multifunctional protein that plays an important role in the development of neural and mesodermal tissue, processes that are regulated by activin-like/bone morphogenetic protein signaling. Bone morphogenetic protein is expressed in the smooth muscle progenitors in the mouse embryonic gut, and bone morphogenetic protein type 2 plays an essential role in smooth muscle differentiation [14]. It is possible that absent or dysfunctional SIP1 contributed to deregulation of the smooth muscle differentiation program which manifested as an additional smooth muscle layer in our patient.
We present the 1st case report of an additional enteric smooth muscle layer in a patient with MWS and Hirschsprung disease. Although the exact etiology is unknown, the abnormality is likely to be a congenital malformation and potentially responsible for the patient's unusual symptom of persistent diarrhea after definitive treatment of Hirschsprung disease. Further work to study the pathology, clinical course, and outcome of Hirschsprung disease in MWS will potentially yield important information in managing these patients.