
Abstract
Respiratory colonization of preterm infants with Ureaplasma urealyticum is a significant risk factor for bronchopulmonary dysplasia, a chronic lung disease characterized by arrest of alveolar development, variable interstitial fibrosis, and disordered elastic fibers in the distal airspaces. As indicated in previous studies, moderate to severe fibrosis is a hallmark of pathology in the Ureaplasma-infected preterm lung. To further characterize the preterm lung's response to Ureaplasma, lung specimens from 4 gestational controls (GC), 12 other pneumonia and 5 Ureaplasma-infected infants were analyzed by immunohistochemistry for α-smooth muscle actin (αSMA) and transforming growth factor β1 (TGFβ1), Hart's elastin staining, and in situ hybridization for tropoelastin (TE) expression. Cells positive for αSMA were observed in thickened, extensive bundles surrounding terminal airspaces in Ureaplasma and other pneumonia cases compared to individual myofibroblasts in GC. The myofibroblast pattern correlated with the severity of fibrosis, but not duration of ventilation. Transforming growth factor β1 immunostaining was primarily localized to alveolar macrophages and was increased in Ureaplasma more than in other pneumonia cases. Elastic fibers and TE-expressing cells were spatially limited to emerging septal tips in GC. In pneumonia cases, increased deposition of elastic fibers was observed surrounding terminal airspaces, but TE expression was similar to GC. In Ureaplasma specimens, accumulation of elastic fibers correlated with duration of ventilation, and TE expression was extensive throughout the walls of terminal airspaces. These findings suggest that Ureaplasma is associated with alveolar macrophage TGFβ1 immunostaining and myofibroblast proliferation contributing to abnormal septation, interstitial fibrosis, and a prolonged and strong elastogenic response in the preterm lung.
INTRODUCTION
Respiratory colonization of the preterm infant with Ureaplasma urealyticum [1–3] is a significant risk factor for the development of bronchopulmonary dysplasia (BPD), a disease characterized by alveolarization arrest, chronic inflammation, variable interstitial fibrosis, and abnormal accumulation of elastin [4–7]. In a review of lung pathology of archived autopsy specimens from Ureaplasma-infected preterm infants, we observed moderate to severe fibrosis in all Ureaplasma-infected infants compared to gestational controls and infants who died with pneumonia from other causes [8]. Ureaplasma may contribute to lung injury and fibrosis by modulating the local immune response to produce sustained chronic inflammation. In the autopsy specimens from Ureaplasma-infected preterm infants, there were increased numbers of tumor necrosis factor (TNF)α-immunoreactive cells in all Ureaplasma-infected infants compared to controls [8]. Infants who are colonized with bacteria or Ureaplasma have higher tracheal aspirate interleukin (IL)-1β concentrations and neutrophil chemotactic activity on day 1 than do noncolonized infants [9]. Tumor necrosis factor a and IL-1β [10] and the profibrotic CC chemokine monocyte chemoattractant factor protein 1 (MCP-1) [11] are elevated in tracheal aspirates during the first few weeks of life in Ureaplasma-infected infants. In a mouse Ureaplasma pneumonia model, intratracheal inoculation with Ureaplasma induces a sustained recruitment of neutrophils and macrophages into the lung [12]. We propose that the interaction of Ureaplasma infection, hyperoxia, and volutrauma contributes to a prolonged proinflammatory, profibrotic response that interrupts normal developmental signaling in the preterm lung.
Normal development of the lung extracellular matrix is critical to the lung's structure and function and is dependent on the spatial distribution and function of myofibroblasts. Myofibroblasts produce collagen to provide structural integrity and limit tissue stretch and elastin to confer elastic recoil to the lung. During the saccular and alveolar stages of lung development, focal elastic fibers are deposited by myofibroblasts at sites where alveolar septation occurs to increase the complexity and surface area of the gas-exchange units [13]. As septa emerge and elongate, elastic fibers remain at the tips of septal ridges and form a network of cables that connect the mouths of alveoli in alveolar ducts and likely facilitate coordinated opening and relaxation of alveoli during respiration [14]. In contrast, elastic fibers accumulate in the BPD lung at sites where septa would normally form in developing lung but the septa fail to emerge and elongate. The malformed and excess elastic fiber bundles at these sites may also be refractile to successful remodeling that would be required for resolution of failed alveolar development. The root causes of excessive and abnormal elastic fiber accumulation associated with BPD are not yet clear but correlate with ventilator-induced lung injury in a sheep model of BPD [15]. Whether Ureaplasma infection contributes to abnormal elastin accumulation in the preterm lung is unknown.
Transforming growth factor β1 (TGFβ1) stimulates myofibroblast differentiation and synthesis of collagen and elastin [13,16,17]. Transforming growth factor β1 has been implicated in lung morphogenesis, repair of lung injury, airway remodeling, and diseases of lung fibrosis [18]. Overexpression of TGFβ1 in the lungs of newborn transgenic mice causes an arrest in lung sacculation, epithelial differentiation, and vascular development [19–21]. Transforming growth factor β1-expressing adenoviral vectors produced similar effects in the newborn rodent lung [18,22]. Transforming growth factor β was detected at sites of lung injury in association with myofibroblast proliferation in lungs of infants dying with respiratory distress syndrome, implicating TGFβ in the preterm lung response to injury [23]. Tumor necrosis factor α, IL-1β, and TGFβ1 are elevated in tracheal aspirates of infants who progress to BPD [24–28], suggesting that chronic inflammatory, profibrotic factors contribute to the pathogenesis of BPD.
We propose that Ureaplasma-induced lung injury and fibrosis are initiated by an acute inflammatory response mediated by TNFα stimulated inflammatory cell recruitment, up-regulation of TGFβ1, induction of lung myofibroblasts, and excessive accumulation of collagen and elastin. In this study, we sought to investigate possible differences in myofibroblast distribution, TGFβ1 expression, elastin accumulation, and tropoelastin expression in human lung specimens from Ureaplasma-infected infants compared to lungs of gestational controls and infants with pneumonias of different etiology. Lung specimens were from the same subject sample as our previous report [8].
METHODS
Subject selection
Preterm infants of 23 to 30 weeks' gestation with autopsy and microbiologically confirmed pneumonia and controls from the same gestational age range who died from nonpulmonary causes were identified from the neonatal intensive care unit database of infants born between January 1992 and March 2000, and their paraffin-embedded lung specimens were obtained from the pathology archives. The lungs were fixed in formalin without attempts to expand the lungs postmortem. The presence or absence of U. urealyticum in each pneumonia case and control was confirmed by polymerase chain reaction (PCR) [8]. The sample included 4 gestational controls who died from nonpulmonary causes, 12 infants with pneumonia who were culture and/or PCR negative for U. urealyticum, and 5 infants with pulmonary disease and positive for U. urealyticum by tracheal aspirate or lung tissue culture or PCR. Clinical and laboratory data of the groups of infants are presented in the previous report [8]. All infants died at 45 d of age or earlier.
Immunohistochemistry
α-Smooth muscle actin and vimentin
Immunohistochemical studies were performed on 5-μm sections of formalin-fixed, paraffin-embedded tissues to detect α-smooth muscle actin (αSMA), a marker of myofibroblasts and smooth muscle, and vimentin, a marker of cells of mesenchymal origin. Sections were deparaffinized and endogenous peroxidase activity was blocked by incubation with 3% H2O2 in 100% methanol. For antigen retrieval, sections were treated in a microwave oven in the presence of saturated lead II thiocyanate for 10 min. Sections were incubated with ready-to-use monoclonal anti-αSMA or anti-vimentin antibody (Biogenex, San Ramon, CA, USA) for 30 min at room temperature and processed using the Biogenex Super Sensitive Detection System® (Biogenex) according to the manufacturer's protocol. Slides were counterstained with hematoxylin.
Transforming growth factor-β1
For TGFβ1 immunohistochemical analysis, we used a polyclonal antibody raised against a peptide at the carboxy terminus of the precursor form of human TGFβ1 that does not cross-react with other TGFβ isoforms. Lung sections were rehydrated, endogenous peroxidase activity was blocked, and the tissues were treated in a microwave oven in the presence of sodium citrate (10 mM, pH 6.0) for 10 min. After blocking with 2% normal goat serum in phosphate-buffered saline (PBS) for 30 min, polyclonal rabbit anti-TGFβ1 (V) antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was added at 1:200 dilution and incubated overnight at 4°C. After rinsing with PBS, biotinylated goat anti-rabbit IgG was applied at 1:200 dilution for 30 min at room temperature. Tissues were then treated with avidin biotin complex for 30 min at room temperature. Color was developed by exposure to 3,3′-diaminobenzidine tetrahydrochloride for 1 min, and tissue sections were counterstained with hematoxylin. A blinded observer (C.-C.J.S.) reviewed the slides.
Hart's elastin staining
Peripheral lung tissue sections (5 μm) were deparaffinized and rehydrated through graded ethanol to PBS and then soaked in 0.25% potassium permanganate solution for 5 min. Slides were cleared in 5% oxalic acid and soaked in resorcin-fuchsin solution (Poly Scientific, Bay Shore, NY, USA) overnight. After washing in water, sections were counterstained with tartrazine (yellow), dehydrated through graded ethanol, cleared in xylene, and mounted.
Quantification of elastic fiber density
Elastic fiber density was quantified with Optimas 3 software (MediaCybernetics, San Diego, CA, USA). After staining with Hart's elastic fiber stain, a minimum of 10 random lung fields per lung specimen at ×200 magnification were captured with the Axiocam digital camera and Openlab software. Hart's staining followed with tartrazine resulted in black elastic fiber staining and yellow tissue. With the Optimas 3 software, the proportion of the area of each image that was black and yellow was determined. Elastin fiber staining associated with blood vessels and airways were excluded. Statistical analysis was performed using the Student's t-test for independent samples, 2-tailed analysis.
In situ hybridization
35S-labeled antisense or sense riboprobes were generated from the human tropoelastin cDNA pHDE-1 [29] that had been linearized for in vitro transcription (Promega, Madison, WI, USA). Sections of peripheral lung fixed in 10% buffered formalin and embedded in paraffin were deparaffinized, rehydrated, and digested with proteinase K. The tissue sections were blocked with triethanolamine and acetic anhydride and hybridized with denatured radiolabeled riboprobe (40,000 cpm/μL) overnight at 60°C. Following hybridizations, tissue sections were subjected to a series of washes including digestion with RNase A, dehydrated, and then exposed to x-ray film overnight to assess signal. After dipping in photographic emulsion (Kodak, Rochester, NY), slides were exposed for 7 to 14 d and then developed and counterstained with hematoxylin and eosin.
RESULTS
Myofibroblast distribution
Lung specimens from infants dying of non–lung-related causes (gestational controls) at 23- to 30-wk postmenstrual age showed a saccular morphology. Cells positive for αSMA were distributed in continuous circular bundles within large arteries and as thin, discontinuous groups of cells along alveolar ducts and at septal tips (Fig. 1A). In contrast, in most Ureaplasma and other pneumonia cases, αSMA immunoreactive cells were distributed in a pattern of thickened clusters of cells often surrounding respiratory bronchioles, alveolar ducts, and alveolar sacs (Fig. 1B,C). The extent of myofibroblast accumulation and percent of lung involvement appeared to be greater in sections of Ureaplasma cases than in sections of other pneumonia cases. There was no apparent association between increased αSMA and number of days of ventilation. The increase in αSMA cells in Ureaplasma-infected infants at 24- to 27-wk gestation was similar whether duration of ventilation was 7 or 45 d. Airway αSMA immunoreactive cells also stained with vimentin, confirming that these cells were myofibroblasts (data not shown).
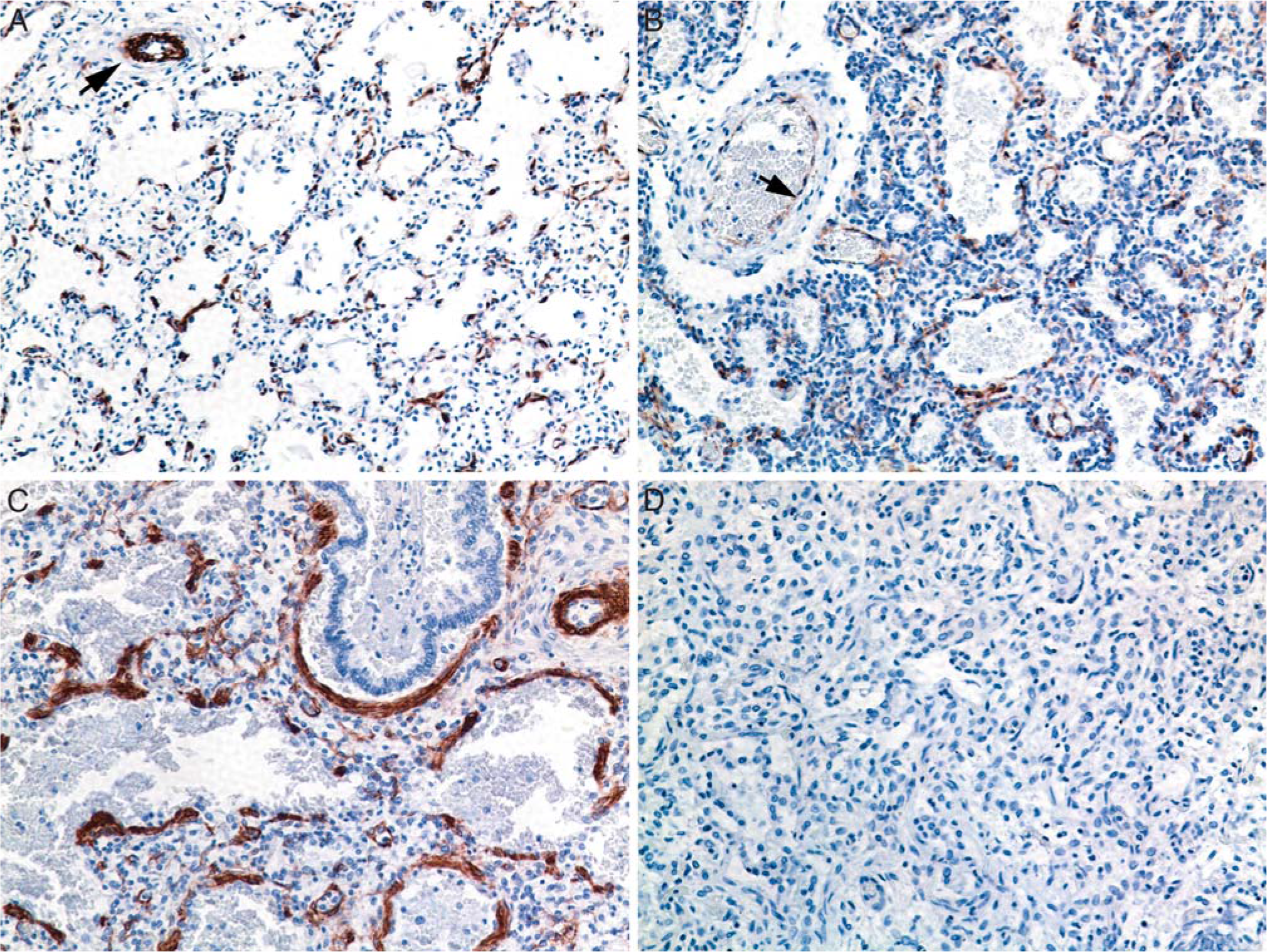
Comparison of α-smooth muscle actin (αSMA) immunostaining in lung specimens.
Transforming growth factor-β1 expression
In gestational controls, there was limited TGFβ1 immunostaining to scattered airway cells, most likely amniotic squamous cells (Fig. 2A). In lung specimens from infants dying with acute bacterial or Ureaplasma pneumonia, immunostaining was concentrated in focal aggregates of alveolar and interstitial macrophages (Fig. 2B,C). In lung specimens from 3 of 4 controls, there were less than 6 TGFβ1 immunoreactive cells/high-power field (hpf). In contrast, there was greater than 6 immunoreactive cells/hpf in specimens from 4 of 12 (33%) other pneumonia cases and 4 of 5 (80%) Ureaplasma cases.
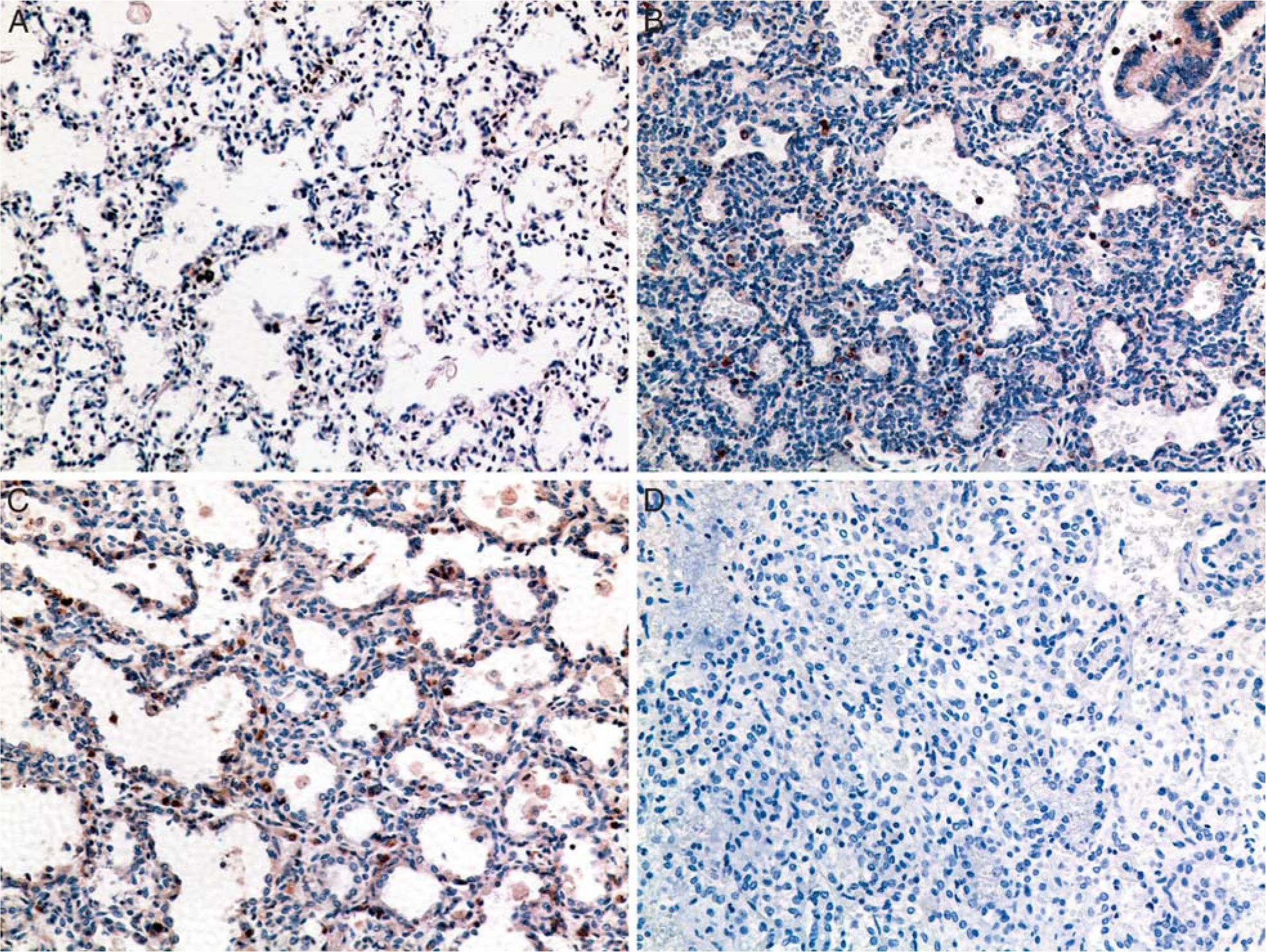
Comparison of transforming growth factor β1 (TGFβ1) immunostaining in lung specimens.
Elastin content and tropoelastin mRNA expression
Elastic fibers, assessed by histochemical staining, were sparse in distal lung specimens from gestational controls (Fig. 3C). The density of elastic fibers in specimens was assessed by color analysis of digitized images of distal lung and expressed as a percentage of total area. In gestational controls, elastic fiber density was low and tropoelastin mRNA, assessed by in situ hybridization, was not readily detected in peripheral lung sections (Fig. 3A,B). In pneumonia specimens with duration of ventilation in the neonatal intensive care unit ranging from 1 to 17 d, accumulations of elastic fibers were apparent in the walls surrounding alveolar ducts (Fig. 3F,I). Elastic fiber density was elevated compared to gestational controls but did not significantly correlate with duration of ventilation (r2 = 0.17, P = 0.26) (Fig. 4). Tropoelastin expression was not elevated above that seen in control specimens (Fig. 3D,E). In the Ureaplasma specimens, accumulations of elastic fibers were commonly found surrounding alveolar ducts and alveolar sacs (Fig. 3I,L). Elastic fiber density was highest in this group and exhibited strong correlation with duration of ventilation (r2 = 0.90, P = 0.015) (Fig. 4). In the Ureaplasma specimens, tropoelastin mRNA was detectable after 20 d of ventilation (Fig. 3G,H) and robust throughout walls of alveolar ducts/sacs after 45 d of ventilation (Fig. 3J,K).
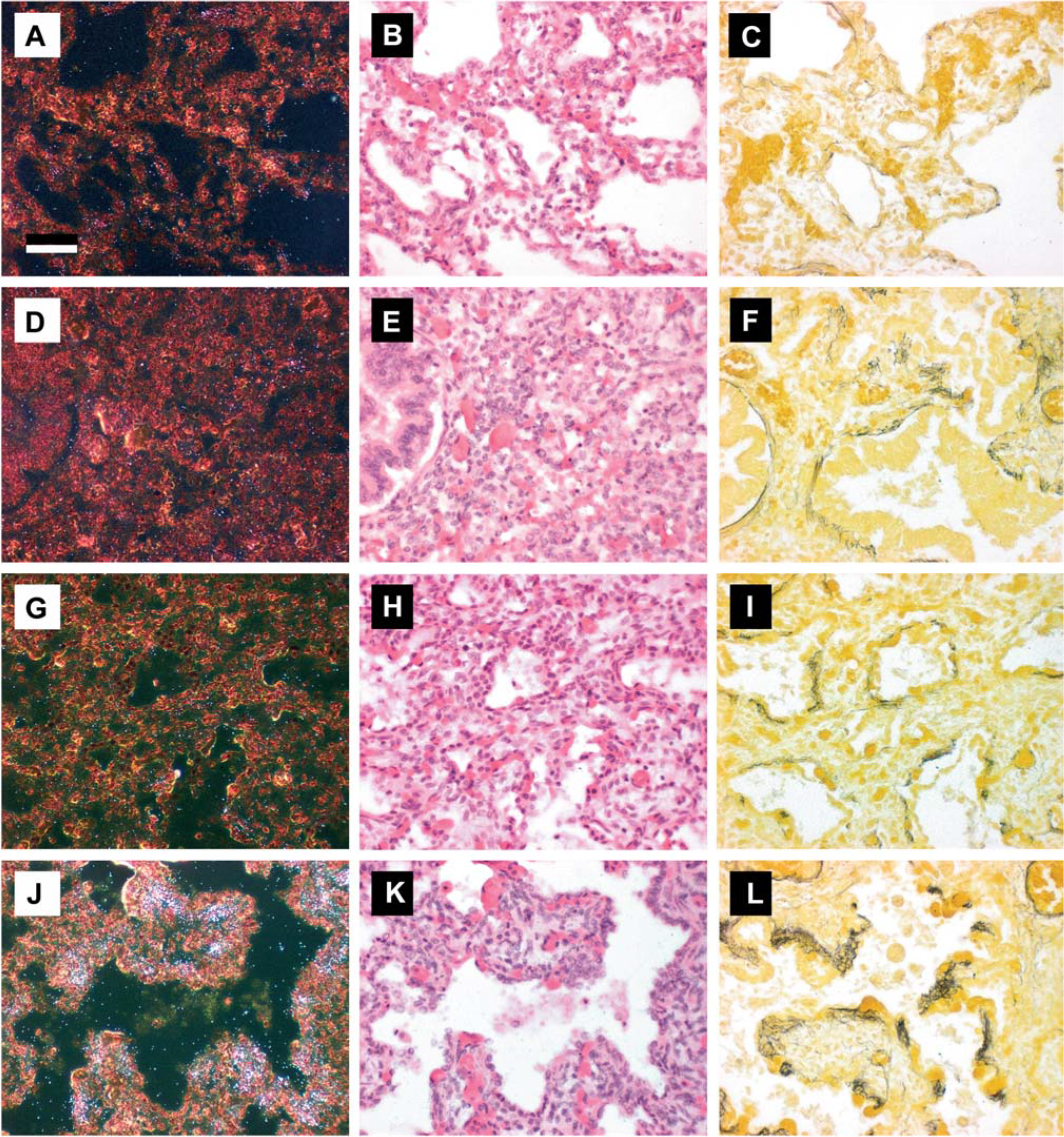
In situ hybridization for tropoelastin mRNA and Hart's resorcin-fuschin stain for elastin in distal lung tissue.
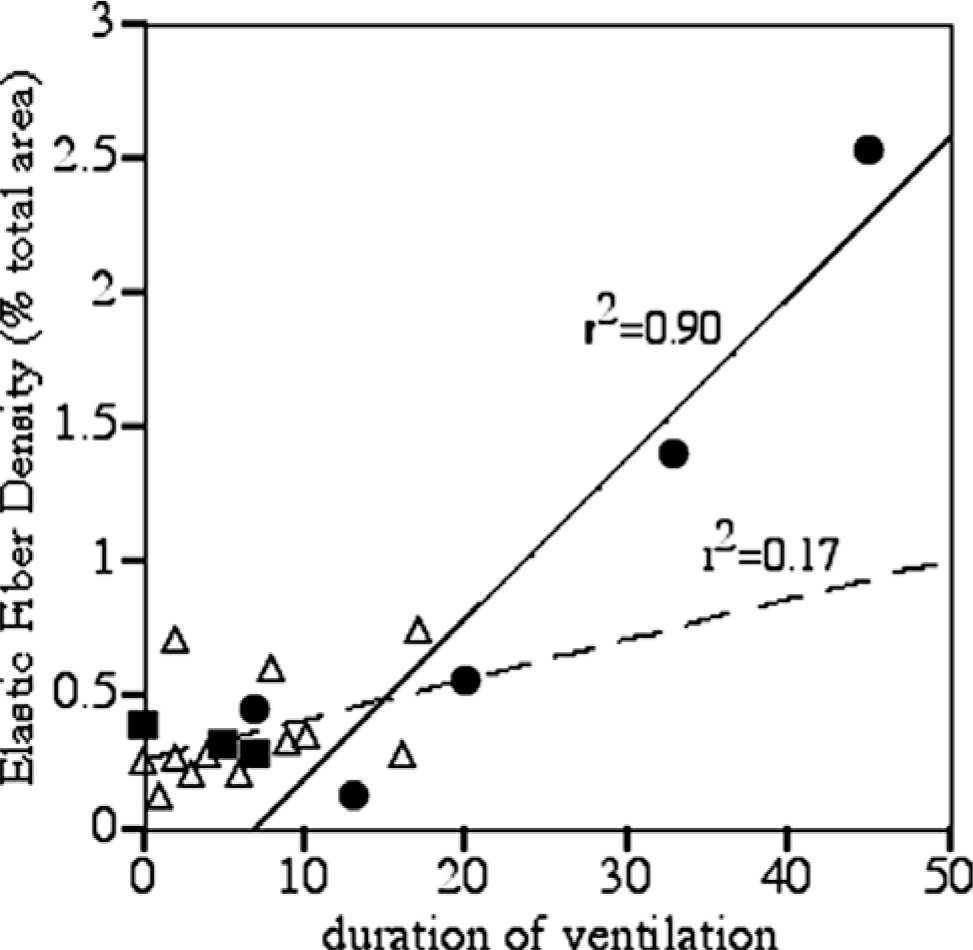
Relationship of elastic fiber density in lung specimens and duration of ventilation. A minimum of 10 random fields per lung specimen was analyzed for the proportion of Hart's elastic fiber stain using Optimas 3 software (MediaCybernetics, San Diego, CA, USA), and the average for each sample was expressed as percent of the total area. The duration of ventilation (x-axis) and elastic fiber density (y-axis) are shown for individual subjects in control (N = 3; filled squares), U. urealyticum pneumonia (N = 5, filled circles), and other pneumonia (N = 11, open triangles) groups. There was a strong positive association between duration of ventilation and elastic fiber density in Ureaplasma pneumonia (r2 = 0.9, P = 0.015) but not in other pneumonia cases.
DISCUSSION
Infants dying with U. urealyticum pneumonia demonstrated marked myofibroblast proliferation, alveolar macrophage TGFβ1 immunostaining, and elastin accumulation. These findings were less pronounced in infants dying with acute pneumonia from other causes. Tropoelastin mRNA expression in other pneumonia infants was no different from gestational controls, but tropoelastin expression was increased in Ureaplasma-infected infants, indicating a prolonged and strong elastogenic response. Although volutrauma and oxygen exposure also contributed to lung injury in these infants, these findings support a role for infection of prenatal or postnatal onset in the development of BPD [1,3,30–33].
Myofibroblasts positive for αSMA are important in normal lung morphogenesis and in response to lung injury [23,34]. α-Smooth muscle actin cells in gestational controls in the present study were primarily found as individual myofibroblasts, consistent with the pattern typical of the saccular stage of lung development. With normal progression of lung development, the number of αSMA cells associated with the acinus decrease [34]. However, with injury of the preterm lung, myofibroblasts increase in number along terminal airways [23]. In Ureaplasma-infected infants, thickened bundles of αSMA cells were localized around respiratory bronchioles and alveolar ducts and persisted up to 6-wk postnatal age, suggesting a prolonged response to infection/inflammation-mediated lung injury.
Transforming growth factor β1 is important in lung morphogenesis and in the response to lung injury. In agreement with the present study, Toti and colleagues [23] observed that TGFβ immunostaining using a non–isoform-specific antibody was localized to alveolar macrophages in preterm infants with evolving BPD. However, Toti and colleagues reported that TGFβ immunoreactive cells were present only in infants dying between 4 and 16 d, and not at later ages. In contrast, in the present study, TGFβ1 immunoreactive inflammatory airway cells were observed in Ureaplasma-infected infants greater than 2 wk of age. The TGFβ1 antibody detects total TGFβ1, so positive staining is not proof of bioactivity. Concurrent immunostaining with anti–phospho-smad2 or -smad3 antibodies to detect TGFβ1 signaling might demonstrate co-localization with TGFβ1 immunostaining [21] but would be difficult to quantitate. However, taken together with our previous observation of persistent inflammatory cell infiltrates in human Ureaplasma pneumonia [8] and in animal models of experimental Ureaplasma pneumonia [12,35], the observations of the present study suggest that Ureaplasma stimulates a chronic inflammatory response in the lung, thereby inhibiting normal developmental septation.
In our previous report, Ureaplasma infection was associated with increase numbers of TNFα-immunoreactive cells [8]. Tumor necrosis factor α overexpression in the lungs of transgenic mice produces enlarged alveolar spaces, thickened alveolar walls, interstitial fibrosis, and endothelial changes [36]. Transient lung overexpression of TNFα by adenoviral gene transfer produces mild lung fibrosis due to TGFβ1 and induction of myofibroblasts [37]. Transient overexpression of IL-1β by adenoviral gene transfer in rat lung stimulates acute inflammation, sequential up-regulation of TNFα and TGFβ1, and development of progressive fibrosis [38]. Both TNFα and IL-1β are elevated in tracheal aspirates in the first weeks of life of infants colonized with Ureaplasma [10]. We speculate that prolonged expression of these cytokines in Ureaplasma-infected infants contributes to increased expression of TGFβ1 and proliferation of myofibroblasts.
The present study demonstrates that Ureaplasma infection of the preterm lung is associated with a robust elastogenic response. An increase in elastin expression and accumulation in the lung could result from an increase in elastin-expressing cells or an increase in elastin expression per cell. Elastin expression in the lung is confined to vascular endothelium and αSMA-positive cells including airway and vascular smooth muscle and alveolar myofibroblasts. In the pneumonia and Ureaplasma specimens, the increased elastic fiber accumulation is likely produced by the αSMA-positive cells in the distal lung. No differences in tropoelastin expression, elastic fiber accumulation, or amount of smooth muscle were noted in the vasculature or proximal airways.
The accumulation of elastic fibers surrounding terminal air spaces in Ureaplasma-infected infants correlated with duration of ventilation as previously observed in the chronically ventilated preterm lamb model [15]. Thibeault and others [7] correlated a respiratory severity score with the volume density and absolute quantity of elastic tissue in human BPD. These authors proposed that elastosis is the preterm lung's response to overstretch. We propose that Ureaplasma infection may augment this response to volutrauma. Notably, as a limitation of this study, the Ureaplasma-positive study group included infants with longer periods of ventilation than the pneumonia or control groups. It is possible that the stronger correlation found in the Ureaplasma group is due in part to the presence of specimens from infants with longer periods of ventilation.
The increase in elastic fiber accumulation and αSMA-positive cells in the distal lung correlated spatially and temporally with the presence of macrophages positive for TGFβ1, suggesting that these are closely linked. Indeed, elastin expression in lung fibroblasts is augmented by TGFβ1 [17]. Thus, TGFβ1 may be a key mediator in this response to infection and mechanical ventilation.