Abstract
Fryns anophthalmia-plus syndrome is a very rare condition initially described by Fryns and colleagues in 1995 in a pair of siblings of nonconsanguineous parents. Since that time, only a few cases have been reported, most of them in newborns and young children. Clinical presentation is variable and includes anophthalmia/microphthalmia, cleft lip/palate, and other facial deformities. Furthermore, skeletal, central nervous system, and endocrine anomalies have been described. We report the case of a male fetus of 18 weeks of gestation with normal karyotype and findings matching Fryns anophthalmia-plus syndrome. Pregnancy was terminated because of sonographically proven facial midline defects and a marked cerebral ventriculomegaly. Macroscopic and histological findings obtained at autopsy showed extreme bilateral microphthalmia, unilateral cleft palate, unilateral nasal deformity, and low-set ears. Skeletal anomalies included 13 pairs of ribs, premature ossification of the calcaneus, and talipes.
INTRODUCTION
As a distinct entity, anophthalmia-plus syndrome was first described by J. P. Fryns and coworkers in 1995 [2]. The index patient was a 17-week-old fetus with bilateral anophthalmia, bilateral cleft lip/palate, macrotia with bilateral facial clefts, large open sacral neural tube defect, and uterus unicornis. The 2.5-year-old brother had bilateral anophthalmia and an absent left ear lobule [2].
We present a fetus with bilateral microphthalmia, unilateral cleft palate, nasal deformity, and minor skeletal anomalies consistent with this syndrome.
CASE REPORT
We report the case of an 18-week-old male fetus of nonconsanguineous parents. The 38-year-old mother (GI/P0) had spontaneous conception with uneventful pregnancy. There was no exposure to teratogens. Chromosome studies on chorionic villus biopsy showed a normal karyotype (46, XY). Prenatal ultrasound revealed facial midline defects and hydrocephalus internus. Therefore, the couple decided to terminate the pregnancy.
At autopsy, growth parameters correlated with a gestational age of 18 weeks (weight 176 g, head circumference 13 cm, crown heel length 19 cm). Facial anomalies included cleft palate with concomitant nasal deformity and macrostomia on the right side (Fig. 1). At 1st sight, the eyes appeared microphthalmical, but at close inspection, bulbi occuli were macroscopically not detectable. Nevertheless, occular structures were histologically demonstrable (Fig. 2). The periorbital structures were macroscopically and radiologically regularly formed. Ears were small and low set with hypoplastic, adherent lobules (Figs. 3,4). Furthermore, there was a posterior rotation of the left ear (Fig. 4). The brain showed considerably dilated ventricles. Fetal radiography displayed 13 pairs of ribs, premature ossification of the calcaneus, and talipes. There were no anomalies of the cardiovascular, gastrointestinal, urogenital, and endocrine systems.

Fetus 18 weeks of age with extreme bilateral microphthalmia, cleft palate on the right side with macrostomia, and deformity of the right nostril.
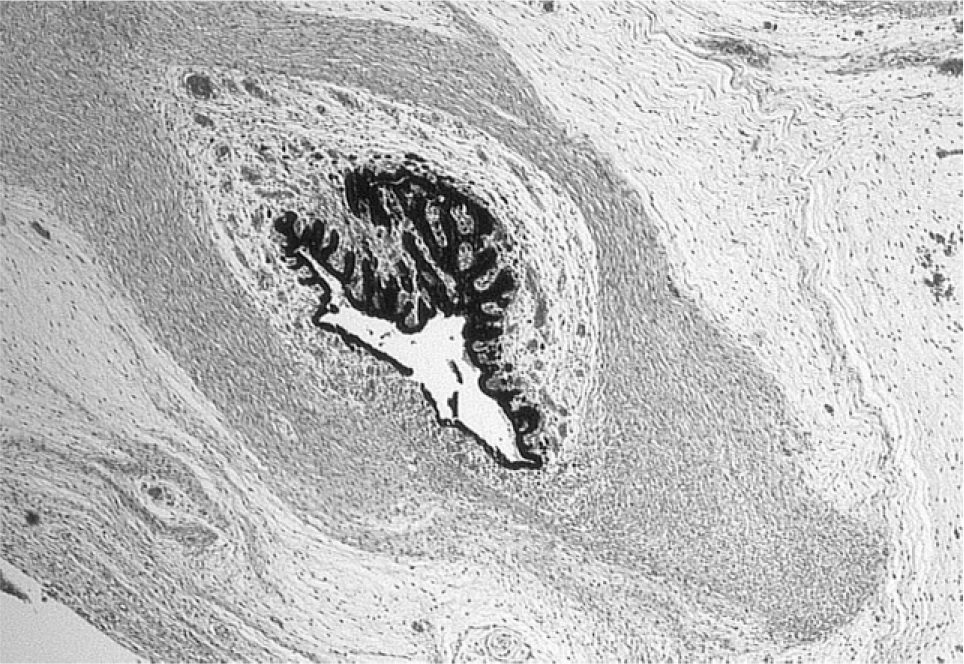
Histological specimen of the microphthalmic eye (H&E).
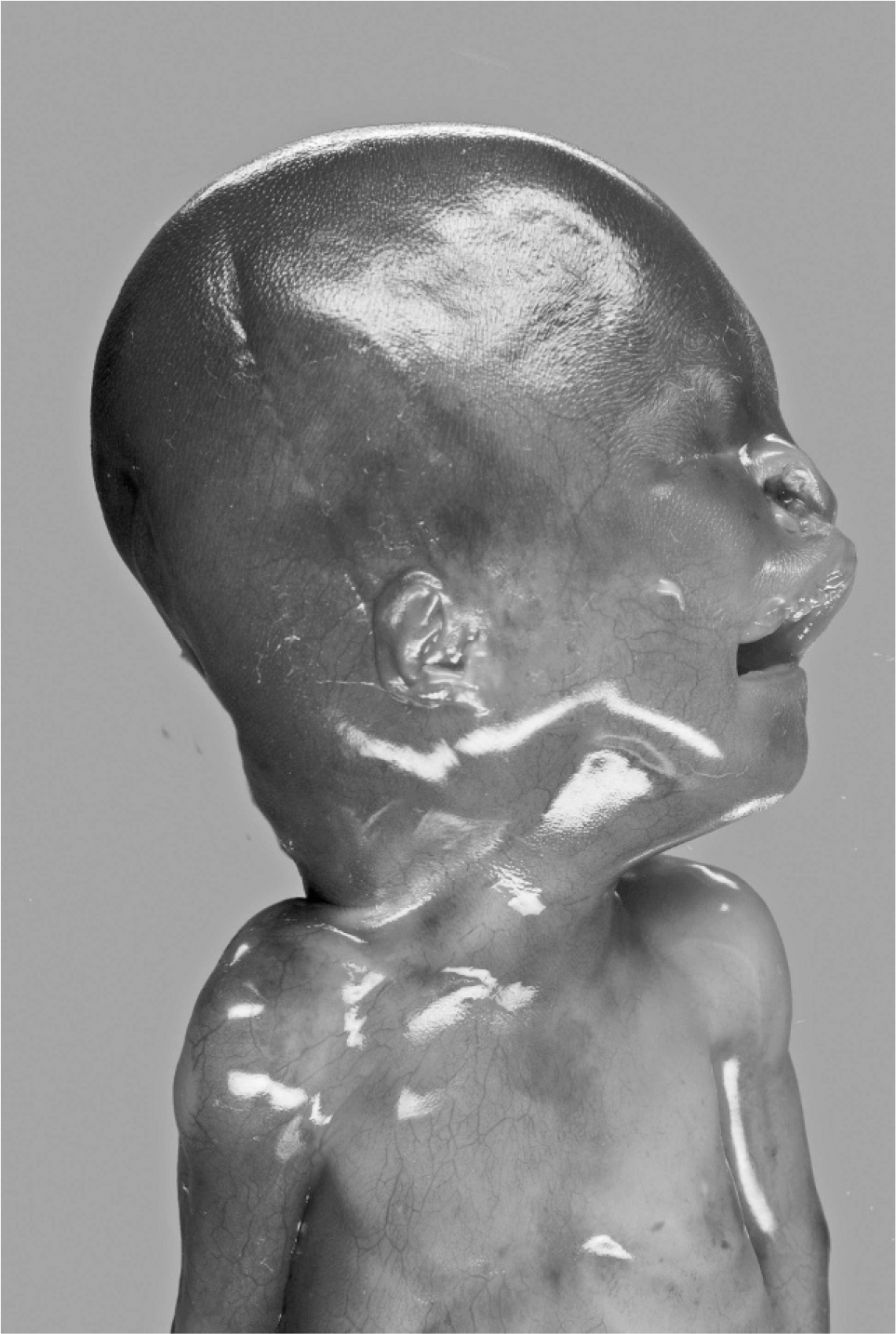
Lateral view: small, low-set ear and hypoplastic, adherent lobule.
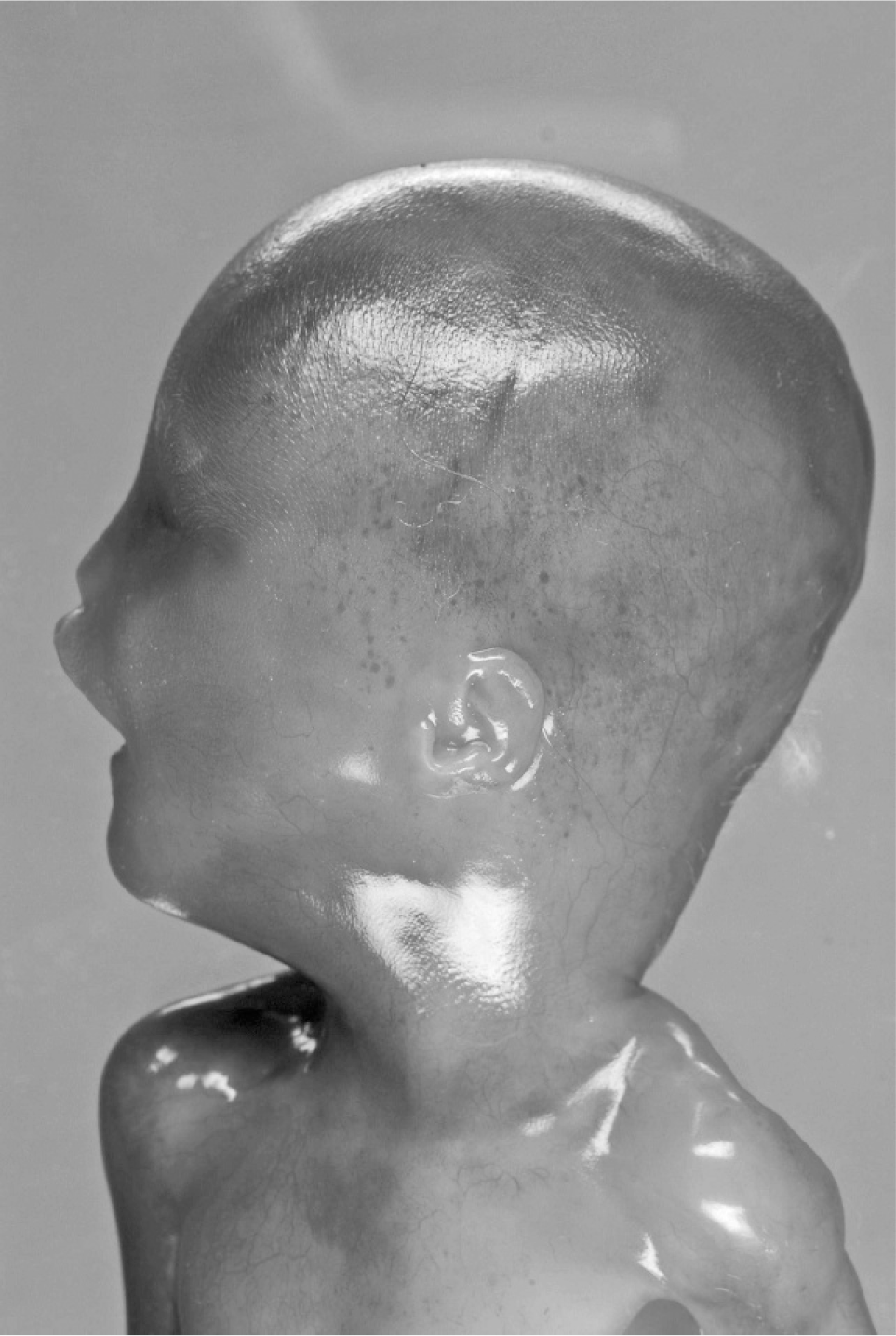
Lateral view (left side): posterior rotation of the left ear.
DISCUSSION
The leading malformations in the present case were microphthalmia and cleft palate, with no other severe internal or external anomalies and no chromosomal aberrations, corresponding to the diagnosis of Fryns anophthalmia-plus syndrome.
Microphthalmia and facial clefts occur in a number of syndromes, such as CHARGE, Lenz microphthalmia, Fraser, and oculo-palato-cerebral syndromes. Frequently, these syndromes are associated with additional anomalies, such as cardiovascular and urogenital malformations. Amniotic bands may also cause deformities in the craniofacial area. In general, facial clefts caused by amniotic bands are asymmetrical and often bizarre and display scar tissue along the cleft. Immunosuppressive medications during pregnancy are also thought to be related to congenital malformations, including microphthalmia and cleft lip/palate [3].
Before Fryns and colleagues published their case of anophthalmia-plus syndrome, 2 other cases of anophthalmia combined with cleft lip/palate and normal karyotype had been described [6,7]. The authors discussed a possible new distinct entity, but it was Fryns who gave this entity a name.
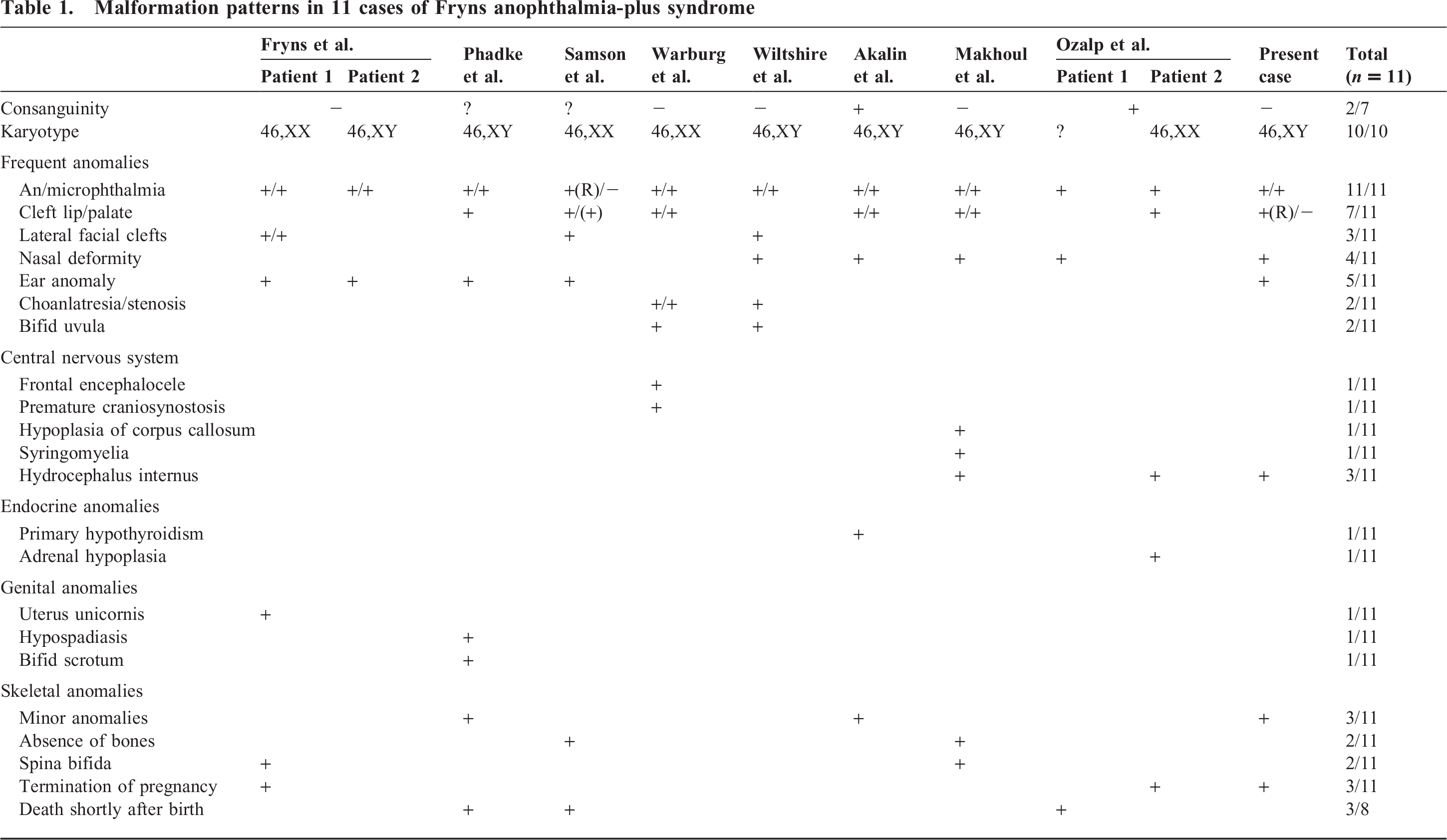
Since then, only a few additional cases have been reported. Inheritance pattern is supposed to be autosomal recessive. Two case reports of consanguineous couples might support this theory [1,5]. The gene locus of anophthalmia-plus syndrome is still unknown. Clinical features vary; malformation patterns of the different patients, including the present case, are listed in Table 1. However, the combination of anophthalmia/microphthalmia, facial clefts, and normal karyotype is a hallmark for this syndrome. Central nervous system and skeletal malformations can be much more pronounced than in our fetus. Makhoul and colleagues [4] presented a patient with multiple vertebral defects and corpus callosum hypoplasia. Two patients with choanal stenosis/atresia and bifid uvula were described [8,9]. Abnormality of endocrine organs may also occur [1,5]. Defects in anophthalmia-plus syndrome can be detected by fetal ultrasonography, which led to pretermination of pregnancy in 3 of 11 published cases. Three of 8 liveborn patients died shortly after birth. The cause of death remained unclear, because autopsies were not performed.
Malformation patterns in 11 cases of Fryns anophthalmia-plus syndrome