
Abstract
We present a case of pediatric anaplastic lymphoma kinase–positive large B-cell lymphoma (ALK-positive LBCL) with a cervical, mesentery, and pelvis cavity mass. Histologic examination of the cervical mass revealed that the lesion was composed of diffuse large immunoblastic-like or plasmablastic-like tumor cells with a sinusoidal growth pattern. The tumor cells were strongly immunoreactive for ALK; revealed a granular cytoplasmic distribution; and were diffusely positive for CD45, CD4, CD138, epithelial membrane antigen, and multiple myeloma oncogene-1 but negative for CD20 and CD79a. The patient underwent 5 courses of cyclophosphamide, doxorubicin, vincristine, prednisone, and etoposid and obtained a remarkable clinical response with regression of mesentery and pelvis cavity mass. We suggest that this distinct subtype of large B-cell lymphoma should belong to the spectrum of pediatric lymphomas and that radiologic examination should be performed to inspect the progression of disease even if the patients experienced complete remission at initial chemotherapy.
INTRODUCTION
Anaplastic lymphoma kinase–positive large B-cell lymphoma (ALK-positive LBCL) is a rare but distinct lymphoma with unusual clinicopathologic characteristics and molecular cytogenetic features that differ from ALK-positive anaplastic large cell lymphoma (ALCL). The 2008 World Health Organization (WHO) classification of lymphomas has been listed as a separate entity under the mature B-cell non-Hodgkin lymphoma and accounts for <1% of diffuse large B-cell lymphoma (DLBCL) [1]. Histologically, this lymphoma shows a sinusoidal growth pattern and is composed of large plasmablastic and/or immunoblast-like cells with round pale nuclei containing large central nucleoli and abundant cytoplasm. The lymphoma cells in most of the reported cases have a unique immunophenotypic profile: they are ALK positive, and they are negative for B- and T-cell lineage markers, with the exception of plasma cell makers and CD4 aberrancy. Since its first report by Delsol and colleagues in 1997 [2], there have been approximately 50 cases reported in the English literature. However, to our knowledge, only 11 ALK-positive LBCLs in children have been reported to date [2–8]. We report the 12th case of a pediatric ALK-positive LBCL and describe the clinicopathologic characteristics of this lymphoma with a review of the other 11 pediatric cases reported in the literature.
MATERIALS AND METHODS
Clinical history
A 17-year-old Chinese female patient presented to a local hospital with a 9-month history of aperiodic abdominal pain. Initially, this manifestation was considered to be gastritis, and the patient was managed with antibiotic administration. Two weeks later, the patient complained that the abdominal pain had become severe and was accompanied by nausea and vomiting. As a result, the patient was referred to our hospital for further examination and treatment. Physical examination revealed that her general condition was stable with normal body temperature and other laboratory results, including blood count, differential, and renal and liver function tests. However, there was an enlarging left cervical mass measuring approximately 4.0 cm in diameter. A computed tomographic scan of the abdomen and pelvis showed that several mass lesions were located in the mesentery and pelvic cavity. The patient was diagnosed with clinical stage III non-Hodgkin lymphoma, and the left cervical mass was surgically resected with no complications. The mass was grey-reddish and well circumscribed to surrounding tissues.
Histology, immunohistochemistry, and in situ hybridization assay
The surgically removed specimen was formalin fixed and paraffin embedded. Hematoxylin and eosin–stained sections were prepared according to standard methods. The immunohistochemistry was performed using the Dako Autostainer with Envision (+) Detection Kit (Dako, Glostrup, Denmark). Immunohistochemical analysis included a broad panel of antibodies against ALK, CD3, CD20, CD30, CD38, CD43, CD45,CD56, CD79a, CD138, kappa, lambda, multiple myeloma oncogene 1 (MUM-1), terminal deoxynucleotide transferase (TdT), pan-cytokeratin, epithelial membrane antigen (EMA), S-100, melanosome (HMB-45), and melan-A. Epstein-Barr virus (EBV) infection was also assessed by in situ hybridization for EBV-encoded small RNA (EBERs) according to the manufacturer's instruction (Dako).
RESULTS
Morphologic features
Histologically, the cervical lymph node showed architecture effacement (Fig. 1A). There was a diffuse infiltration of large tumor cells with a sinusoidal growth pattern (Fig. 1B). Some large tumor cells had an immunoblastic-like appearance with single prominent nucleoli and abundant cytoplasm (Fig. 1C). Some tumor cells had a plasmablastic appearance (Fig. 1D), but atypical multinucleated neoplastic giant cells were absent in the tumor. Hemorrhage and necrotic areas were not found.
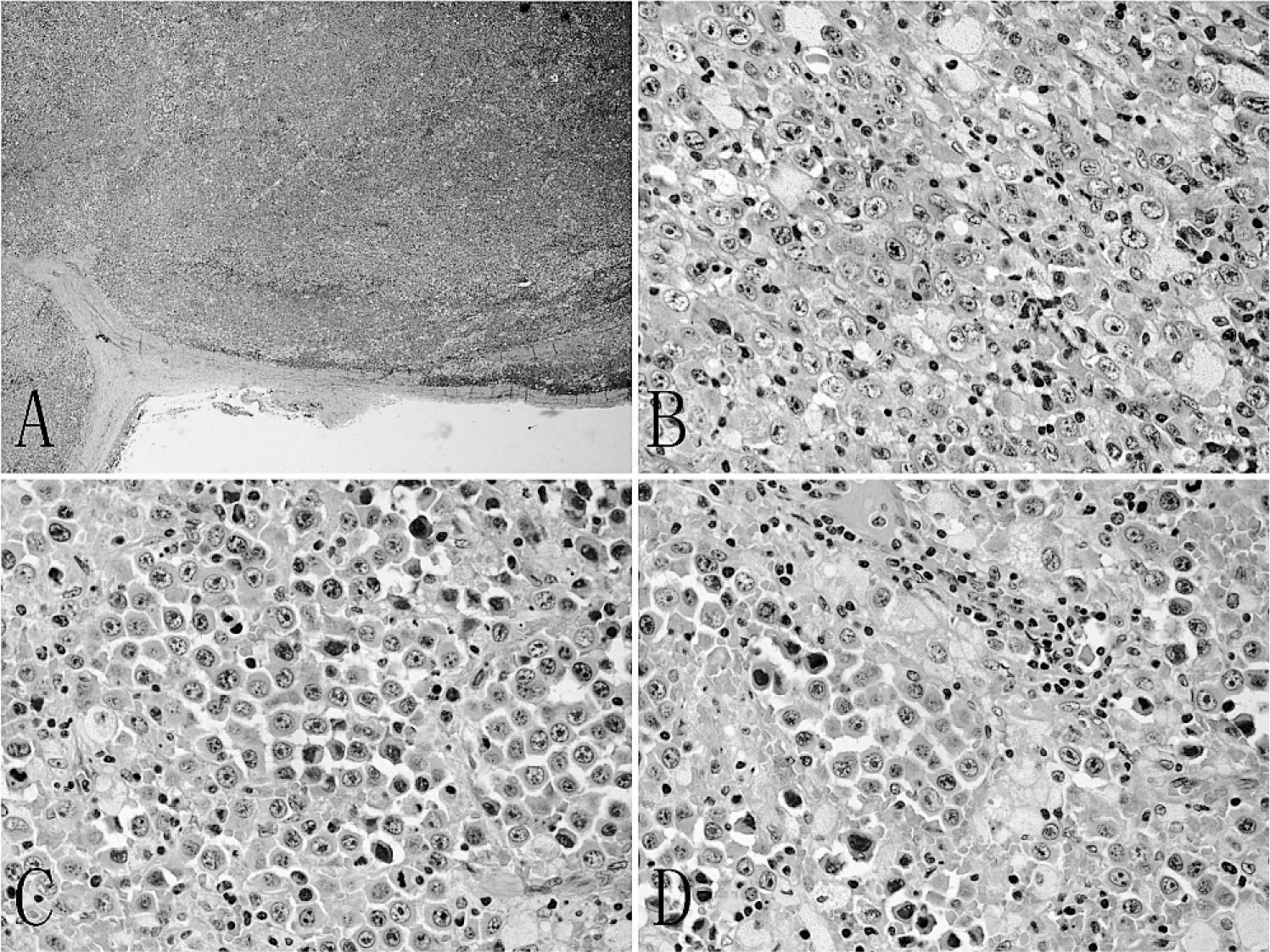
Morphologic features of the left cervical mass.
Immunohistochemical features
Immunohistochemistry showed that the large tumor cells were strongly positive for ALK in a granular cytoplasmic distribution, which has been described in clathrin/ALK-fusion protein cases (Fig. 2A). In addition, the tumor cells showed strong expressions of CD45, CD4, CD138, lambda, EMA, and MUM-1, as well as focal expressions of cytoplasmic IgA and CD30 (Fig. 2B–G). The tumor cells lacked the expression of B-cell markers CD20 and CD79a (Fig. 2H). CD2, CD3, CD7, CD8, CD38, CD43, CD56, CD68, kappa, granzyme B, TdT, pan-cytokeratin, S-100, HMB-45, and melan-A were all negative. The tumor cells were also negative for EBERs by in situ hybridization assay.

Immunohistochemical profile of tumor cells. The tumor cells were strongly cytoplasmic granular positive for anaplastic lymphoma kinase (
Clinical features
On the basis of histopathologic and immunohistochemical findings, the patient was diagnosed with ALK-positive LBCL according to 2008 WHO classification criteria. The patient then received cyclophosphamide, doxorubicin, vincristine, prednisone, and etoposide (E-CHOP regimen) for 5 cycles and achieved a partial response. There was partial regression of the abdominal lymphadenopathy during treatment. After completion of 4 months of chemotherapy, there was neither relapse nor systemic symptoms reported after 6 months of follow up.
DISCUSSION
Anaplastic lymphoma kinase–positive large B-cell lymphoma is a rare subtype of LBCL. It was first described as “large B-cell lymphoma expressing ALK kinase” by Delsol and colleagues in 1997 [2]. In 2001, the WHO classification of tumors of hematopoietic and lymphoid tissue recognized it as a distinct subtype of DLBCL [9]. Histologically, ALK-positive LBCL is characterized by an immunoblastic or plasmablastic microscopical appearance with round nuclei, prominent central nucleoli, and eosinophilic cytoplasm. Sinusoidal growth pattern of tumor cells and atypical multinucleated neoplastic giant cells may be observed frequently. Immunohistochemically, the tumor cells exhibit ALK-positive staining with a granular cytoplasmic pattern in most cases. The staining pattern is highly correlated with cytogenetic abnormality of t(2;17)(p23;q23) or clathrin/ALK fusion [3,5–7,10]. However, few cases showed cytoplasmic and nuclear ALK staining pattern, which was associated with classic ALCL-related t (2; 5) (p23;q35) or nucleophosmin (NPM)/ALK fusion [4,11,12]. In previous 11 pediatric ALK-positive LBCLs, only 2 cases showed NPM/ALK fusion [4]. In the present case, the tumor cells showed a strongly ALK-positive signal in a granular cytoplasmic pattern, which strongly suggested that molecular cytogenetics abnormality was t (2;17) (p23;q23) involving clathrin on 17q23 and ALK on 2p23, although we had not assayed the genetic abnormalities by fluorescent in situ hybridization analysis. In addition, most ALK-positive LBCLs express epithelial membrane antigen and plasmacytic differentiation markers, such as CD138, VS38c, monotypic cytoplasmic light chain, and MUM1, whereas other B-cell–specific markers, in particular CD20 and CD79a, are not expressed. However, cases of ALK-positive LBCL with strong CD20 or CD79a expression have been occasionally described accounting for 11% and 18%, respectively, of the total number of cases [8]. These unique immunophenotypic profiles of tumor suggest that ALK-positive LBCL is derived from postgerminal B-cell lymphocytes that have undergone plasmacytic differentiation. In the present case, microscopic examination revealed that the tumor consisted of larger immunoblastic or plasmablastic cells with ALK (+), CD138 (+), EMA (+), and MUM-1(+). On the basis of these findings, this case was diagnosed as ALK-positive LBCL.
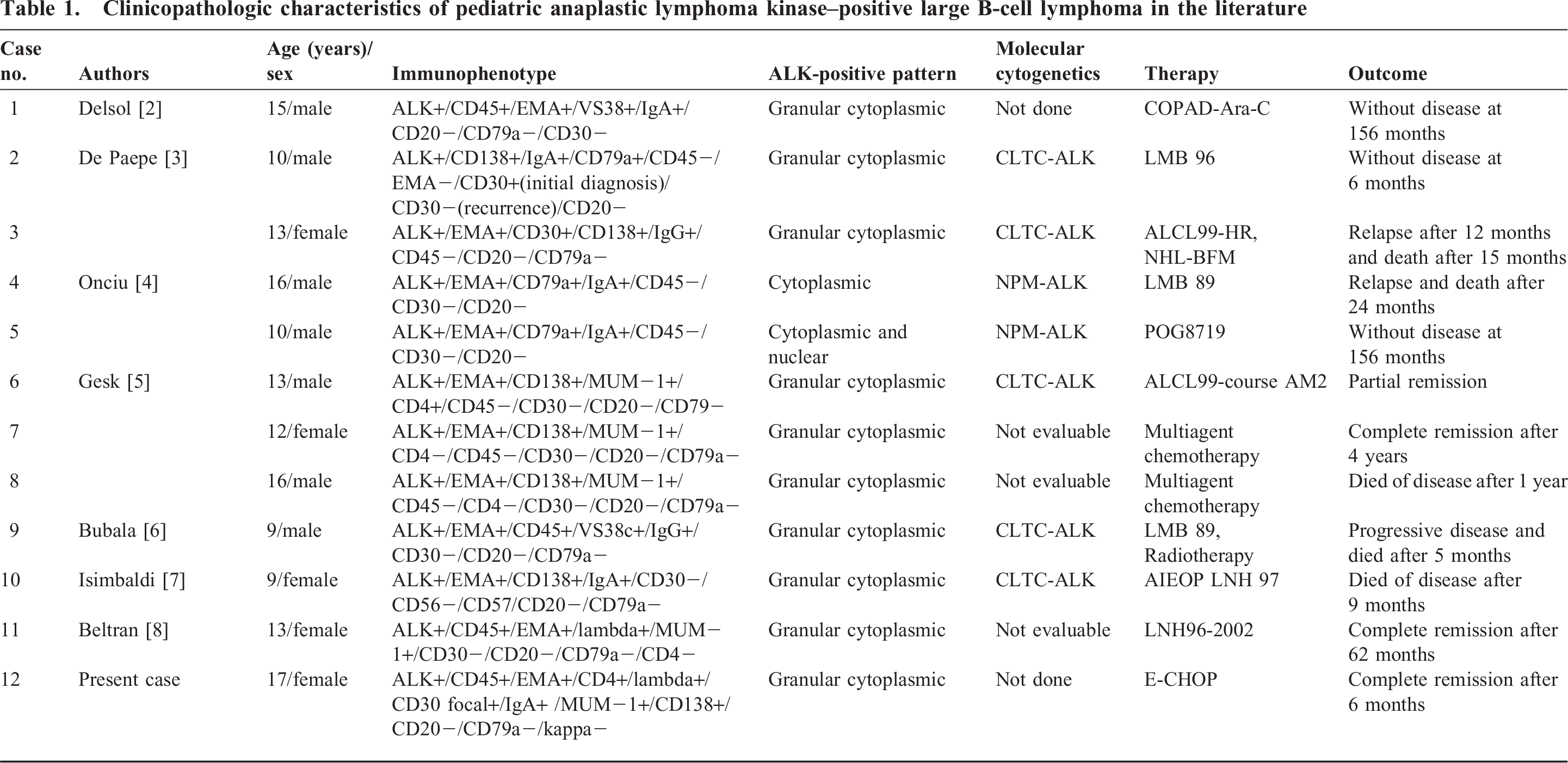
We reviewed the 50 cases of ALK-positive LBCL reported in the English literature between 1997 and 2011, and only 12 cases have been reported in pediatric population [2–8], including our present case, accounting for 23.5% (12/51) of the total number of cases (Table 1). It reveals that ALK-positive LBCLs predominately occur in male adults (male:female 4.3:1) but has a roughly equal gender distribution in pediatric patients (male:female 1.4:1), even though there was no statistical difference between the overall survival of male compared with female patients [8]. With regard to age distribution, a bimodal age curve could be observed in ALK-positive LBCL, showing a peak in children and a second peak in middle-aged adults. In pediatric patients, the average age of presentation was 12.7 years (range 9–17 years), whereas, in adults patients, it was 43.4 years (range 20–72 years). The spectrum of lymphoma in children and young adults is rather limited with 4 main subgroups: Burkitt lymphoma, lymphoblastic non-Hodgkin lymphoma, ALCL, and Hodgkin lymphoma [13]. The age range of ALK-positive LBCL is 9–70 years (median 36 years) [1]. However, we found that almost a quarter of reported ALK-positive LBCL occurred in children. Therefore, ALK-positive LBCL differs somewhat from other subtypes of DLBCL, and this distinction suggests that the tumor should belong to the spectrum of pediatric lymphomas. The most commonly affected areas of ALK-positive LBCLs were cervical and mediastinal lymph nodes. Infrequently, the extranodal sites could be involved, such as bone (8 cases), liver and spleen (4 cases), gastrointestinal tract (3 cases), and nasopharyngeal region (2 cases). In children, all of the reported cases showed enlarged cervical lymph nodes at diagnosis or during the follow-up period.
Clinicopathologic characteristics of pediatric anaplastic lymphoma kinase–positive large B-cell lymphoma in the literature
The main histologic differential diagnoses include ALK-positive ALCL, anaplastic variants of DLBCL, plasmablastic lymphoma (PBL), and primary effusion lymphoma (PEL). These diseases exhibit either ALK-positive immunophenotypic profile (ALK-positive ALCL) or plasmacytic differentiation with CD20 negativity (anaplastic variants of DLBCL, PBL, and PEL), which may sometimes cause diagnostic confusion with ALK-positive DLBCL. Anaplastic lymphoma kinase–positive ALCL shows usually large tumor cells with abundant cytoplasm and pleomorphic nuclei and strong expression of CD30 on the cell membrane and in the Golgi region. However, CD30 is usually negative in ALK-positive LBCL, although focal and weak staining has been reported in a few cases of ALK-positive LBCL [3,14], including our present case. Similarly, large round or polygonal cells appearance and CD30 and CD20 positivity would support the diagnosis of anaplastic variants of DLBCL. Plasmablastic lymphoma is a CD20-negative DLBCL with immunoblasts and obvious plasmacytic differentiation resembling plasmablastic plasma cell myeloma. Plasmablastic lymphoma is associated with human immunodeficiency virus (HIV) and EBV and tends to present with extranodal involvement, usually in oral and gastrointestinal sites. Primary effusion lymphoma is another CD20-negative DLBCL, which is universally associated with human herpesvirus 8 (HHV-8) and tends to present in body cavities (eg, pleura, peritoneum). The presence of ALK expression and other plasmacytic markers, such as CD138, VS38c, and MUM-1 and a lack of virus infection (HIV, EBV, and HHV-8), should be helpful in making a diagnosis of ALK-positive LBCL.
The ALK-positive LBCL appears to have a poor prognosis, with 11 months of survival for adults and children [15]. However, some children have enjoyed longer survival (at least 2 cases longer than 156 months) [2,4]. Of the total 50 reported cases in the literature, the strongest prediction factor associated with survival was clinical stage at presentation. Although more intensive therapies in the pediatric population have been found in some reports, there is no difference in survival between pediatric and adult cases [8]. To date, any clinical trial to find an optimal treatment regimen for ALK-positive LBCL has not been reported. Although Crizotinib (PF-02341066), a small-molecule inhibitor of the ALK tyrosine kinase, has shown a therapeutic potential for non–small-cell lung cancer and inflammatory myofibroblastic tumor in early-phase clinical trials [16,17], it has not yet been identified as a targeted therapy for ALK-positive LBCL. Because the tumor cells of ALK-positive LBCL are usually negative for CD20 antigen, this tumor is thus insensitive to rituximab. A case of ALK-positive LBCL has been reported to receive therapy with rituximab, but that patient died 6 months after diagnosis [10]. In pediatric patients with ALK-positive LBCLs, including our present case, it seemed that all of the regimens (LMB 89, LMB96, POG8719, and CHOP) were highly intensive, and only 5 patients (41.7%, 5/12) died of relapse and progressive disease at the time of report [3–7]. In our case, the patient was at stage III and showed sensitivity to 5 cycles of E-CHOP therapy without any sign of relapse and systemic symptoms during the period of follow up. A longer follow-up period and laboratory examination is still necessary for predicting the prognosis of this patient. However, combined with the remainder of the adult cases, standard CHOP regimen seems inadequate to treat ALK-positive LBCL given evidence of progressive disease and multiple recurrences. More intensive therapies or agents active in novel pathways in combination with chemotherapy are necessary for this rare and distinct lymphoma.
Herein we reported a rare case of ALK-positive LBCL occurring in a child. In pediatric patients, the average age of presentation is in the 1st decade. Immunohistochemical staining for ALK, CD20, and CD30 expression and molecular cytogenetic analysis for CLTC-ALK or NPM-ALK fusion are useful in the differential diagnosis between ALK-positive LBCL and other lymphoma with plasmacytic differentiation. Because ALK-positive LBCL appears to have an aggressive biologic behavior with poor prognosis, we suggest that a long period of follow up is necessary, even if the patient had a complete remission at initial chemotherapy.