
Abstract
Brown-Vialetto-Van Laere syndrome (BVVLS) is a rare degenerative neurological disorder characterized by pontobulbar palsy and sensorineural deafness. Since its initial description in 1894, fewer than 100 cases have been reported, and published neuropathological analyses of these cases are extremely rare. Recently, individuals with BVVLS have been found to carry mutations in the C20orf54 gene, which encodes the human homolog for a rat riboflavin transporter. We present the case of a male who presented at the age of 5 years with sensorineural deafness, as well as those of 2 infant sisters who presented at 11 and 13 months of age with weakness and ataxia, respectively. All cases were genetically confirmed. We include the 1st immunohistochemical characterization of C20orf54 expression in BVVLS and controls. Results showed punctate axonal staining in the control cases that was dramatically reduced in the 3 BVVLS cases compared to the 5 controls. This decreased staining was seen even in the neocortex, which was unaffected in the BVVLS cases by routine histology. While the implications of these results are far from definitive, and although the evaluation of more cases is needed, immunohistochemistry for the C20orf54 protein may eventually be useful, in the right clinical scenario, as a screening test when selecting cases for sequencing of the C20orf54 gene to diagnose BVVLS at autopsy.
INTRODUCTION
Brown-Vialetto-Van Laere syndrome (BVVLS) is a rare neurological disorder first described by Brown in 1894 [1] as familial amyotrophic lateral sclerosis (ALS) and then later described by Vialetto in 1936 [2] and Van Laere in 1966 [3]. Brown-Vialetto-Van Laere syndrome is characterized by progressive pontobulbar palsy and sensorineural deafness. The age of onset varies from infancy to early adulthood, with an average onset in the 2nd decade of life. Early-onset cases tend to be more severe. Patients usually present with cranial nerve VII–XII palsies with or without hearing loss. Occasionally the hearing loss precedes the other neurologic signs. In rare severe cases, affected individuals do not develop deafness, presumably because they expire before they have the chance to develop the symptom. Other initial presenting symptoms include muscle/limb/facial weakness, respiratory compromise, and slurring of speech. As the disease progresses, the development of long tract degeneration, cranial nerve III–IV palsies, cerebellar ataxia, and lower motor neuron signs give rise to a clinical picture similar to that associated with ALS [1–5]. About half of reported cases are sporadic, the other half following mostly an autosomal recessive pattern of inheritance. Clinically, the differential diagnosis includes other conditions with lower cranial nerve and upper/lower motor neuron signs, such as ALS (adult presentation, no deafness), Fazio-Londe syndrome (no deafness), and Madras motor neuron disease. The latter condition is almost clinically indistinguishable from BVVLS, differing only in its being more commonly sporadic (85% versus 50% in BVVLS), lacking in cranial nerve III–IV dysfunction [6], and missing newly discovered genetic mutations [7].
Recently, genetic analyses of a consanguineous family with multiple affected individuals with BVVLS have demonstrated mutations in the C20orf54 gene, which encodes the human homolog of a rat riboflavin transporter and is thought to play a role in riboflavin transport in human as well [7,8]. This latter fact has led to the use of riboflavin supplementation as a treatment to the disorder, with the apparent effect of cessation of symptom progression [9,10]. To date, fewer than 100 cases have been reported, mostly as clinical case reports in the neurology literature. Series with detailed neuropathological analysis of these cases, most of which involve young children, are very rare [6–11].
The reported pathologic findings usually include neuronal injury/loss and white matter tract degeneration, predominantly involving the brain stem and cerebellum. Specific affected areas/findings described include cranial nerves and cranial nerve nuclei III–XII, neuronal loss and gliosis in the substantiae nigrae, loci cerulei, inferior colliculi, subthalamic nuclei, solitary nuclei, cuneate and gracile nuclei, olivary nuclei, loss of cerebellar Purkinje neurons, loss of anterior horn spinal motor neurons, and degeneration of the spinocerebellar, central tegmental, solitary, and pyramidal tracts as well as the medial longitudinal fasciculus. The neurons of the neocortex, allocortex, and deep gray matter structures are spared [9–13].
In this article we present 3 cases of BVVLS with genetically confirmed mutations in the C20orf54 gene and outline their neuropathological features. In addition, we present the results of applied immunohistochemistry using a commercially available C20orf54 immunostain, which has not been previously attempted on cases of BVVLS.
CASES
Patient 1
The 1st patient was a 22-year-old man diagnosed with BVVLS at the age of 12. There was no previous family history of neurological disease and the parents were not consanguineous. He presented at the age of 5 years with sensorineural deafness, followed by the progression of speech difficulties and weakness. Recurrent bouts of respiratory difficulties eventually led to a tracheostomy and consistent positive pressure ventilation. His weakness progressed to quadriplegia and his neurologic status continued to deteriorate. He was eventually only able to communicate with a “brain fingers” device and finally only with eye movements, until he lost that ability to communicate a year before his death in 2011 from sepsis.
Patients 2 and 3
Patients 2 and 3 were sisters who were born without complications. There was no previous family history of neurological disease and the parents were not consanguineous. Patient 2 had some delay of motor milestones but presented at the age of 11 months with ptosis, neck weakness, and respiratory distress. She progressed to flaccid quadriplegia and required full life support within 4 months of the onset and died at 25 months of age. It was thought that she had a hearing deficit as well. Patient 3 was born 15 months after patient 2 and presented at the age of 13 months with a subacute onset of irritability and ataxia. Cerebrospinal fluid analysis revealed transitory elevation of protein. Neurophysiological studies revealed attenuated sensory responses and sensorineural deafness. She demonstrated sensory ataxia and weakness that was milder than her that of her sister. She began to have severe respiratory difficulties, and her care was withdrawn 6 months after the onset.
METHODS
C20orf54 immunostaining
Paraffin sections from brain stem and frontal neocortex were stained with the C20orf54 antibody (Santa Cruz, polyclonal rabbit, 1∶200 dilution) using Bond Epitope Retrieval solution 2 (ER 2, Leica) as an antigen retrieval step. Initially the immunohistochemistry protocol was developed using normal surgical biopsy material of the human salivary gland (submandibular) as a positive control. Subsequent positive controls included age-similar specimens from the brains of both surgical and autopsy cases. For the negative control experiment, we ran a preabsorption study in which the antibody was mixed with the antigenic peptide used to generate the antibody, with the objective of eliminating the binding of the antibody to the protein in the tissue. An aliquot of the primary antibody was incubated with a 100-fold molar excess of the antigenic peptide resuspended in distilled water with 2 mmol/L dithiothreitol for 30 minutes with gentle agitation at room temperature. All immunostaining procedures were performed on a Benchmark XT automated stainer (Ventana). Omission of the primary antibody was performed as a negative reagent control and resulted in no detectable staining.
RESULTS
A summary of the clinical, pathologic, and genetic findings can be found in Table 1. Immunohistochemical results of C20orf54 staining of controls can be found in Figure 1.
Summary of clinical, pathologic, and genetic findings of the 3 Brown-Vialetto-Van Laere syndrome (BVVLS) cases

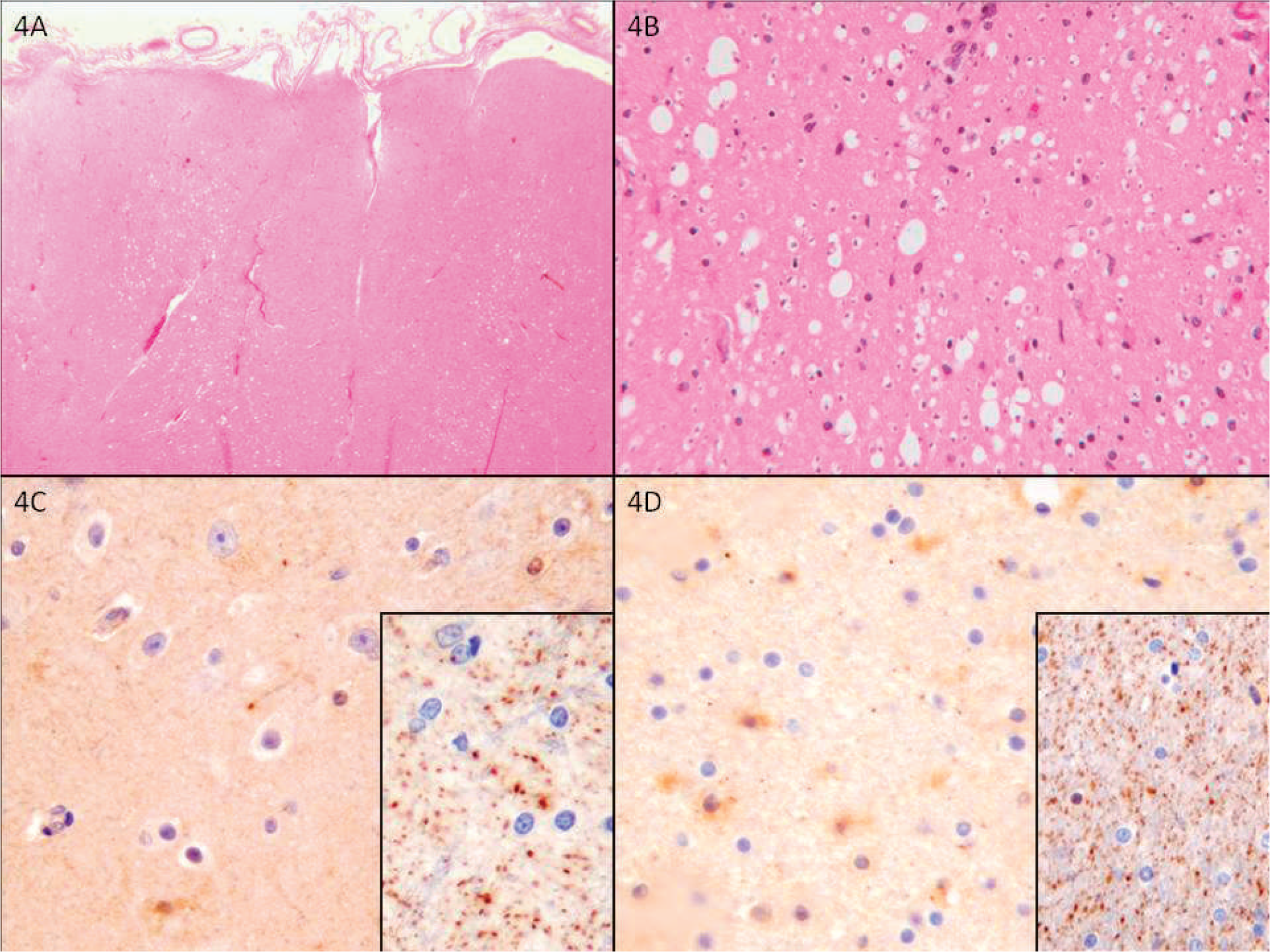
C20orf54 immunostain of neocortical gray matter in (
Case 1 (Fig. 2)
Case 1: (
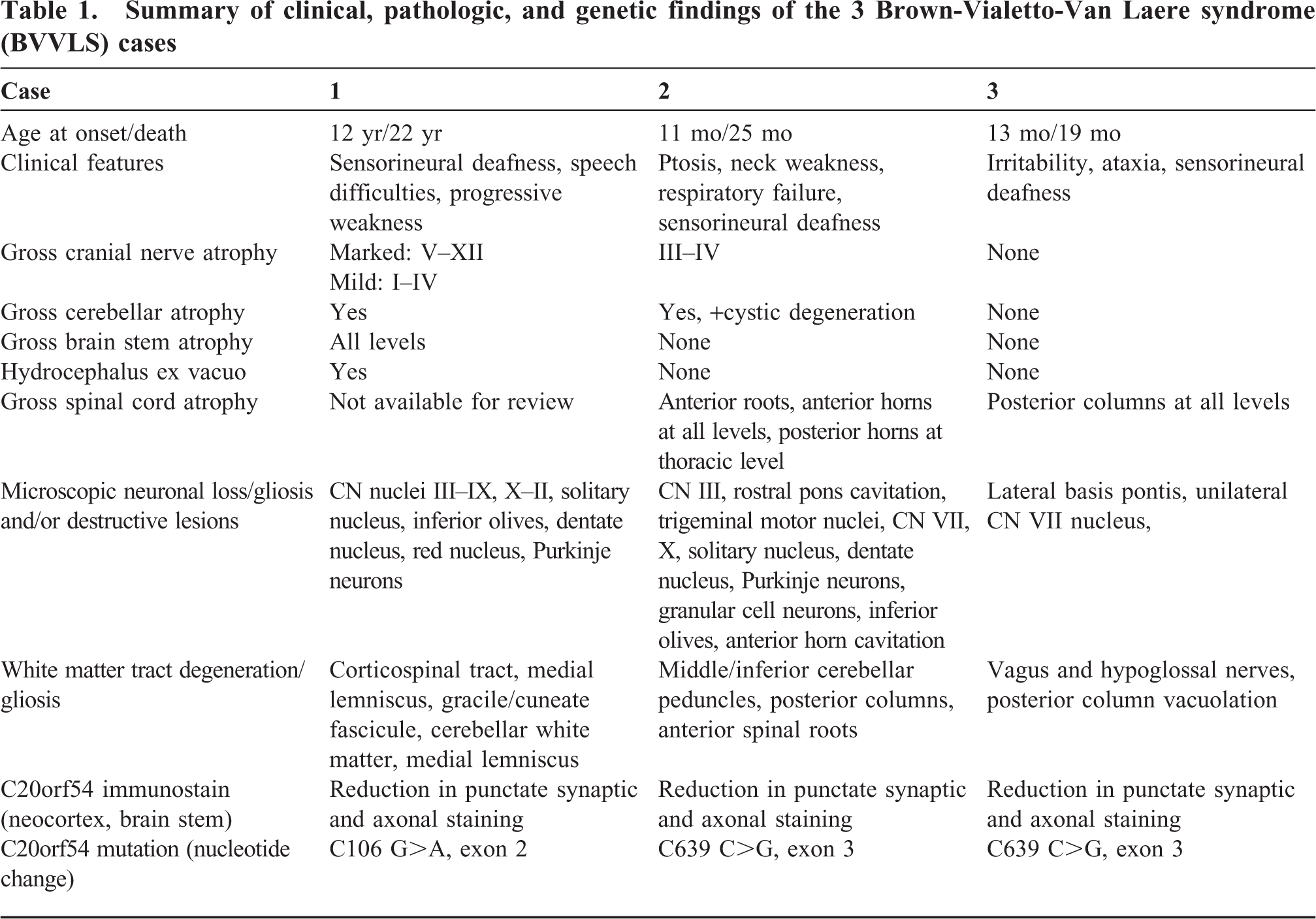
Only the brain was available for examination. The brain weighed 1378 g prior to fixation. Gross examination was significant for severe atrophy of the pons, medulla, and cranial nerves V–XII, with moderate atrophy of the midbrain and lesser atrophy of cranial nerves I–IV. Coronal sectioning revealed marked hydrocephalus ex vacuo (Fig. 2A). Transverse sectioning of the brain stem and cerebellum demonstrated diffuse gray discoloration of the pons and medulla (Fig. 2B) as well as of the cerebellar white matter and dentate nuclei, respectively (Fig. 2C).
On routine hematoxylin and eosin staining there was a near-total loss of neurons in the inferior olivary nuclei, dentate nuclei of the cerebellum, medullary reticular formation, red nuclei, nuclei of cranial nerves V–XII (Fig. 2D), and neurons of the basis pontis (Fig. 2E). The corticospinal tracts showed marked vacuolation. Sections of the cerebellum were significant for moderate loss of Purkinje neurons with associated Bergmann gliosis (Fig. 2F) and severe rarefaction of the subcortical and deep white matter.
Luxol Fast Blue staining of sections of the rostral and caudal medulla revealed a generalized, near-total loss of myelin in all white matter tracts. A Hirano silver stain and internexin immunostain showed a near-total (>90%) loss of axons in the medullary corticospinal tracts and medial lemniscus bilaterally. Sections of the neocortex did not reveal any significant pathologic changes. Immunohistochemistry for TDP-43, polyglutamine, alpha-internexin, alpha-synuclein, phospho-tau (AT8), and ubiquitin were performed on sections of the brain stem and neocortex. No abnormal staining or inclusions were identified. Immunostaining with the C20orf54 antibody on sections of the brain stem, frontal neocortex (Fig. 2G), and deep white matter (Fig. 2H) demonstrated marked reduction in punctate synaptic and axonal staining.
Case 2 (Fig. 3)
Case 2: (
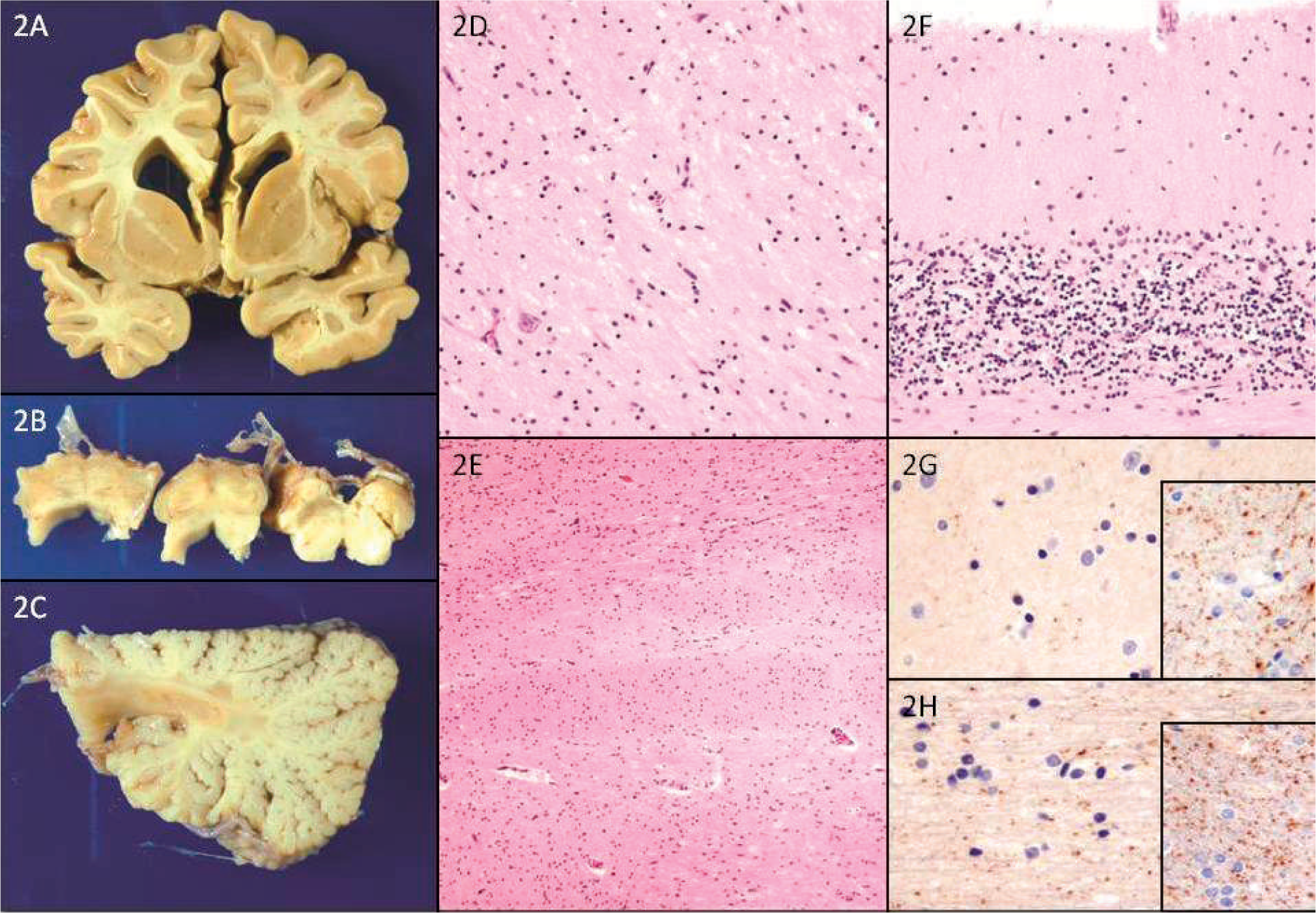
The brain and distal spinal cord were available for examination. The brain weighed 1050 g after fixation. There was no atrophy of the forebrain, but the cerebellum was smaller than normal. The oculomotor and trigeminal nerves appeared grossly atrophic, but the optic nerves were intact. Coronal sections of the cerebral hemispheres demonstrated no evidence of ventricular enlargement and no focal abnormalities. Transverse sections of the brain stem showed no atrophy of the pyramidal tracts or cerebellar peduncles. Sagittal sections of the cerebellum demonstrated cortical atrophy with sulcal prominence. The lateral cerebellar hemispheric white matter showed dark discoloration and a granular, firm texture with several cystic areas measuring 3–7 mm in diameter bilaterally. The spinal cord had severe atrophy of the anterior roots, and transverse sections revealed brown discoloration of the anterior horns at all levels with involvement of the posterior horns in thoracic levels.
On microscopic sections the significant changes were confined to the brain stem, cerebellum, and spinal cord. There was dense gliosis and neuronal loss in the midbrain tegmentum involving, but not confined to, the oculomotor nuclei. The rostral pons had bilaterally symmetrical zones of partial cavitation with border gliosis and macrophages with neuronal loss, rarefaction, and gliosis in other areas of the tegmentum, including the trigeminal motor nuclei, lateral basis pontis, and middle cerebellar peduncles. These bilaterally symmetrical changes extended throughout the pons, but there also were more gliotic changes involving the facial nuclei and radicles of the facial nerves. The medulla had gliotic areas with neuronal loss involving the hypoglossal nuclei, dorsal motor nuclei of the vagi, and the nuclei of the solitary tracts (Fig. 3A). The inferior olivary nuclei had near-total neuronal loss with dense gliosis. The inferior cerebellar peduncles displayed subacute destructive lesions with macrophages, similar to what was seen in the pons. The caudal medulla showed subacute destructive lesions that were mild in the dorsal column nuclei and more severe in central portions (Fig. 3B–D). The descending motor fibers were spared at all levels of the brain stem. The subacute partially cavitary lesions extended into the deep cerebellar white matter bilaterally, but there also was gliotic rarefaction of other portions of the white matter. The dentate nuclei had intense gliosis and neuronal loss, and the cerebellar cortex had severe loss of granular neurons and mild to moderate loss of Purkinje cells and reduction of the thickness of the molecular layer (Fig. 3E). Surviving Purkinje cells frequently had axonal torpedoes and occasionally had dilated dendrites. The cerebellar atrophy was most severe in the anterior and superior regions of the hemispheres. The spinal cord had cavitary loss of gray matter with near-complete loss of anterior horn neurons. At the lumbar levels the changes were less cavitary but had rarefaction with neuronal loss and gliosis. The anterior roots were severely atrophic. Posterior horns also were affected but the roots were relatively spared. The posterior columns had axonal loss but the corticospinal tracts were spared. Immunostaining with the C20orf54 antibody on sections of the brain stem and neocortex (Fig. 3F) demonstrated marked reduction in punctate synaptic and axonal staining compared with adult and age-similar pediatric control cases.
Case 3 (Fig. 4)
Case 3: (
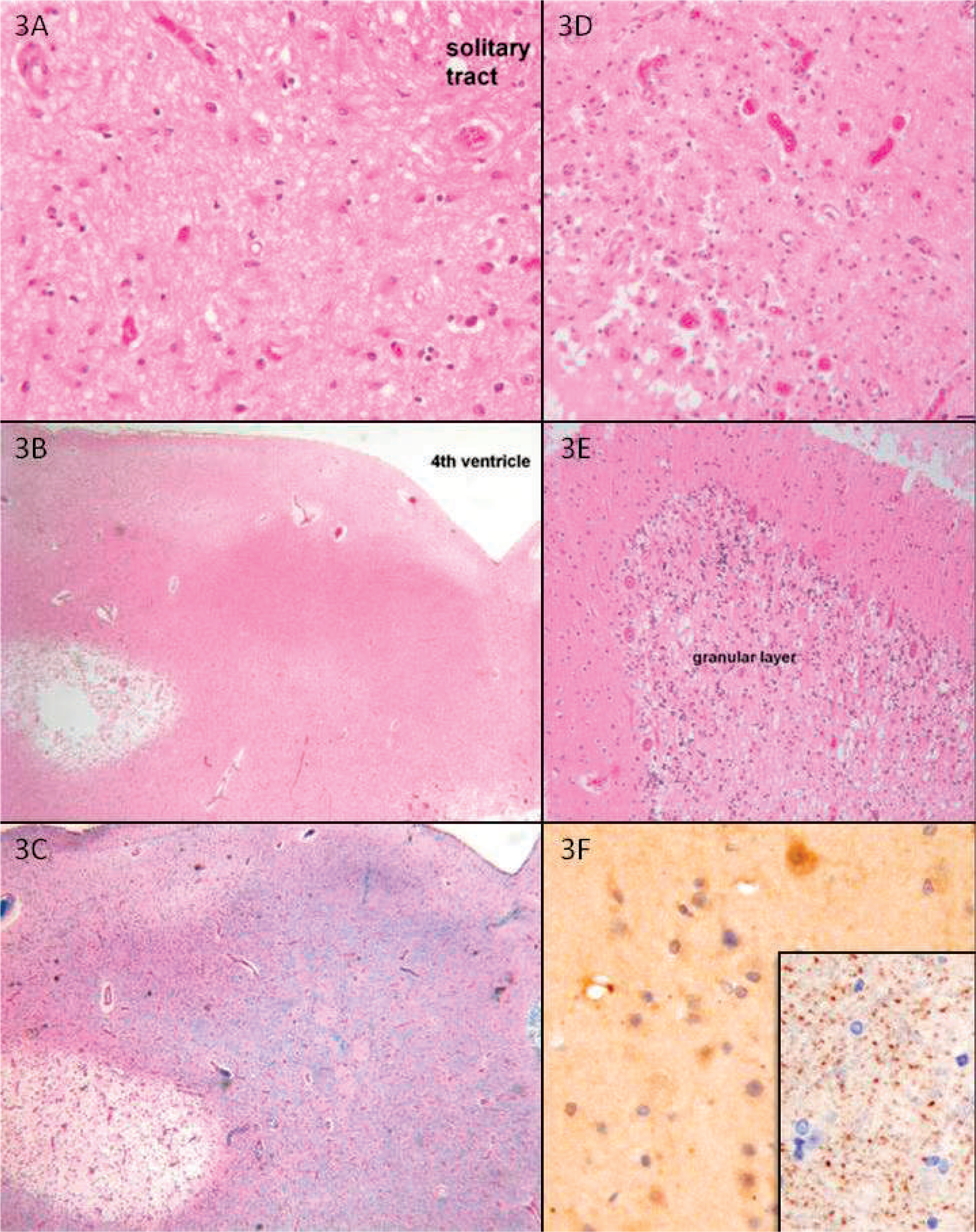
The brain and spinal cord were available for examination. The weight of the brain after fixation was 1250 g. The brain was slightly swollen, with mild notching of the anterior unci but no obvious herniation. The cranial nerves were grossly intact. Coronal sections of the cerebral hemispheres were unremarkable, as were transverse sections of the brain stem and sagittal sections of the cerebellum. Transverse sections of the spinal cord showed gray discoloration of the posterior columns at all cervical levels.
Microscopic sections of forebrain were unremarkable. Microscopic sections of brain stem had a few patches of vacuolation in the lateral pons with rare foamy macrophages. There was slight vacuolation and mild gliosis in one facial nucleus. There was mild loss of axons in the vagus and hypoglossal nerves but no significant changes in the corresponding nuclei. The cerebellar cortex and deep nuclei were unremarkable. Sections of the spinal cord had vacuolar changes with mild gliosis and infiltration of foamy macrophages in the posterior columns (Fig. 4A,B), most pronounced at cervical levels but present throughout the cord (). There was minimal gliosis in the anterior horns at the cervical level without evident anterior or posterior root atrophy or neuronal loss. Immunostaining with the C20orf54 antibody on sections of the brain stem, neocortex (Fig. 4C), and subcortical white matter (Fig. 4D) demonstrated marked reduction in punctate synaptic and axonal staining compared with adult and age-similar pediatric control cases.
DISCUSSION/CONCLUSION
We have presented 3 cases of genetically confirmed BVVLS with what is, to our knowledge, the 1st attempt to immunohistochemically target the protein product of the recently discovered mutated gene. It is interesting to note both the clinical/pathologic similarities and differences among our cases as well as those between our cases and those previously reported. The patient in case 1 presented in the classic fashion, with sensorineural hearing loss, while the patients in cases 2 and 3 presented with weakness/respiratory failure and irritability/ataxia, respectively. The clinical course for patient 1 was prolonged, while those of patients 2 and 3 were more acute and precipitous, consistent with their early presentation. All 3 exhibited sensorineural deafness.
Pathologic findings for all patients were similar to those described in the literature, demonstrating varying degrees of gross atrophy and microscopic neuronal loss and white matter tract degeneration, mainly confined to the brain stem and cerebellum. The degree of the gross atrophy, neuronal loss/gliosis, and the geographic extent of the areas affected was much greater in patient 1, given his prolonged disease course, while patients 2 and 3 demonstrated more areas of spongy/cystic degeneration. Similar to previous reports, the neuronal populations of the neocortex and deep gray matter nuclei were grossly and microscopically unaffected. Despite the latter finding, however, the C20orf54 immunohistochemical staining was reduced in all 3 cases in sections of the neocortex and subcortical white matter compared to staining in age-matched normal controls. This finding was not anticipated. Initially only brain stem sections were stained, but we could not rule out the possibility that the lack of C20orf54 staining in those sections was secondary to the marked neuronal loss and white matter tract degeneration, rather than a primary finding related to the disease. The finding of reduced staining in the grossly and microscopically unaffected neocortex supports the latter position.
Patients 2 and 3 shared the same genetic mutation in the C20orf54 gene, which differed from the mutation discovered in patient 1 (although his had been reported previously in association with BVVLS [8]). No one has yet correlated the individual mutation variants with clinical severity and disease course, but it may be that different mutations in the C20orf54 gene have variable effects on the disease onset, progression, and severity.
In conclusion, BVVLS is a rare inherited neurodegenerative syndrome that exists in the literature largely in the form of clinical case reports. We have provided detailed clinical and neuropathological descriptions of 3 cases. Each case harbored a mutation of the C20orf54 gene, as previously described in BVVLS patients [8]. In addition, this is the 1st account of immunohistochemistry for the C20orf54 on genetically proven cases of BVVLS. Although studies of additional cases are required for definitive conclusions, the results suggest that in the right clinical setting, immunohistochemistry for the C20orf54 protein may eventually serve as a screening tool when selecting cases for sequencing of the C20orf54 gene to diagnose BVVLS at autopsy. Little is yet known about the function or importance of the genetic mutations or about their relation to riboflavin transport, although at least 1 study [10] has claimed riboflavin supplementation is able to stabilize and inhibit symptom progression. Loss of the normal punctate synaptic/axonal staining may be a primary consequence of the genetic mutation or may be related to secondary axonal degeneration, although the finding of such a pattern in the apparently unaffected cerebral cortex would argue for the former. Brown-Vialetto-Van Laere syndrome is undoubtedly a complex genetic syndrome with a heterogeneous clinical presentation, variable rate of progression, and mixed neuropathologic findings, most of which are likely affected by numerous clinical and genetic factors.
Further questions involve the relation, if any, of the location of the mutation within the gene to the clinical presentation and symptom severity; the function of the gene in the human brain; and its relationship to the cause of neurodegeneration. Such advances will only germinate from the reporting and collation of genetically determined BVVLS cases.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank Ping Shang for her help with the development and application of the C20orf54 immunostain, Chan Foong for his assistance with gross dissection and photography, and Kerri Phillips and Katy Bartush for their assistance with the preparation of microscopic sections.