
Abstract
Synovial sarcoma originating in the liver is extremely rare, and thus far only 3 cases have been reported in the English literature. Herein, we report a primary hepatic synovial sarcoma in a 13-year-old Chinese boy. This patient present with a 10-day right upper quadrant pain, and a heterogeneous mass was documented in the right hepatic lobe by computed tomography. Subsequently, the patient underwent right hepatectomy. Histologically, the tumor exhibited classic features of monophasic synovial sarcoma. The diagnosis was confirmed by the presence of SS18 gene rearrangement and identification of SS18-SSX1 fusion transcript. Unfortunately, a relapsing mass was detected 11 months after the surgery. To the best of our knowledge, the current case is the 1st published example in the pediatric population.
Keywords
CASE REPORT
A 13-year-old Chinese boy was referred to a local hospital with a 10-day intermittent right upper quadrant pain. Ultrasonography and abdominal computed tomography revealed a heterogeneous mass in the right hepatic lobe beneath the capsule, measuring 8.6 × 5.3 cm (Fig. 1). The preoperative differential diagnosis was between neoplasm and parasite infection. Subsequent needle biopsy of the mass revealed that the lesion was predominantly composed of hyperchromatic spindled cells, highly suspicious for spindle cell malignancy. Subsequently, the patient underwent right hepatectomy in December 2011.
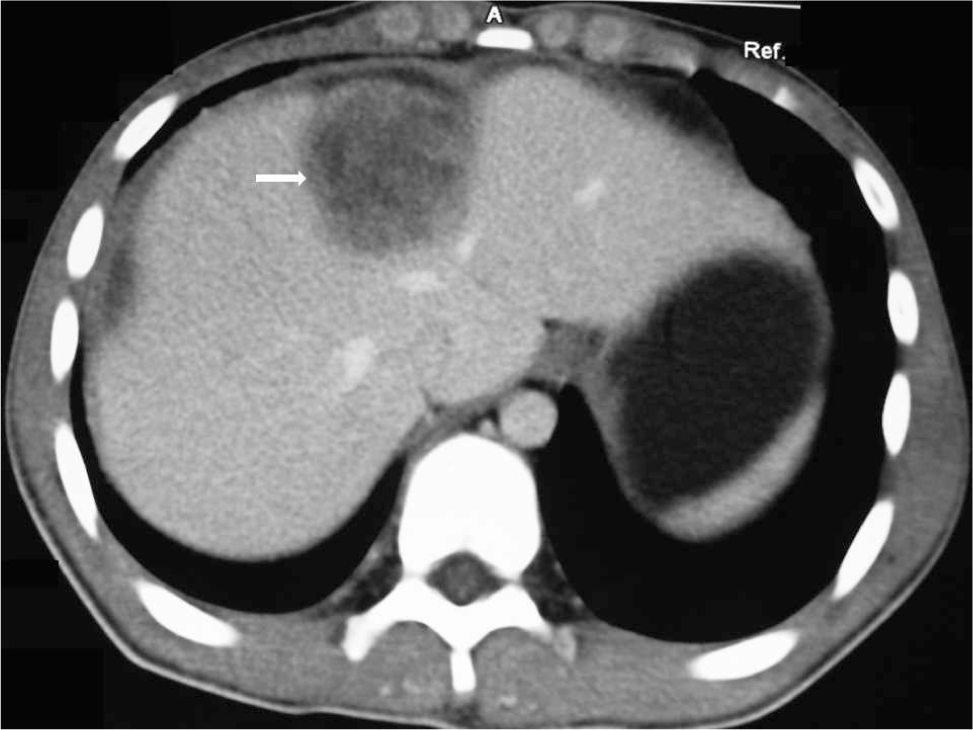
Computed tomographic scan with enhancement revealing a large heterogenous mass arising from the right hepatic lobe (arrow).
This study was approved by the West China Hospital Institutional Review Board. Our department received the consultation slides from the peripheral hospital. Microscopy of the resected specimen showed identical morphology to that of needle biopsy. The lesion was unencapsulated and compressed the adjacent hepatic parenchyma (Fig. 2A) but did not invade the hepatic capsule. The neoplastic cells were arranged in fascicular pattern, and a hemangiopericytoma-like vascular pattern was observed in some areas (Fig. 2B,C). The neoplasm was composed of monomorphous spindle-shaped cells with moderate cytoplasm and hyperchromatic nuclei with inconspicuous, small nucleoli (Fig. 2D). The mitotic activity was frequent (up to 25/10 high-power fields). Focal necrosis and hemorrhage were also noted. No evidence of clear epithelial differentiation was identified. Stromal calcification and ossification were not found.
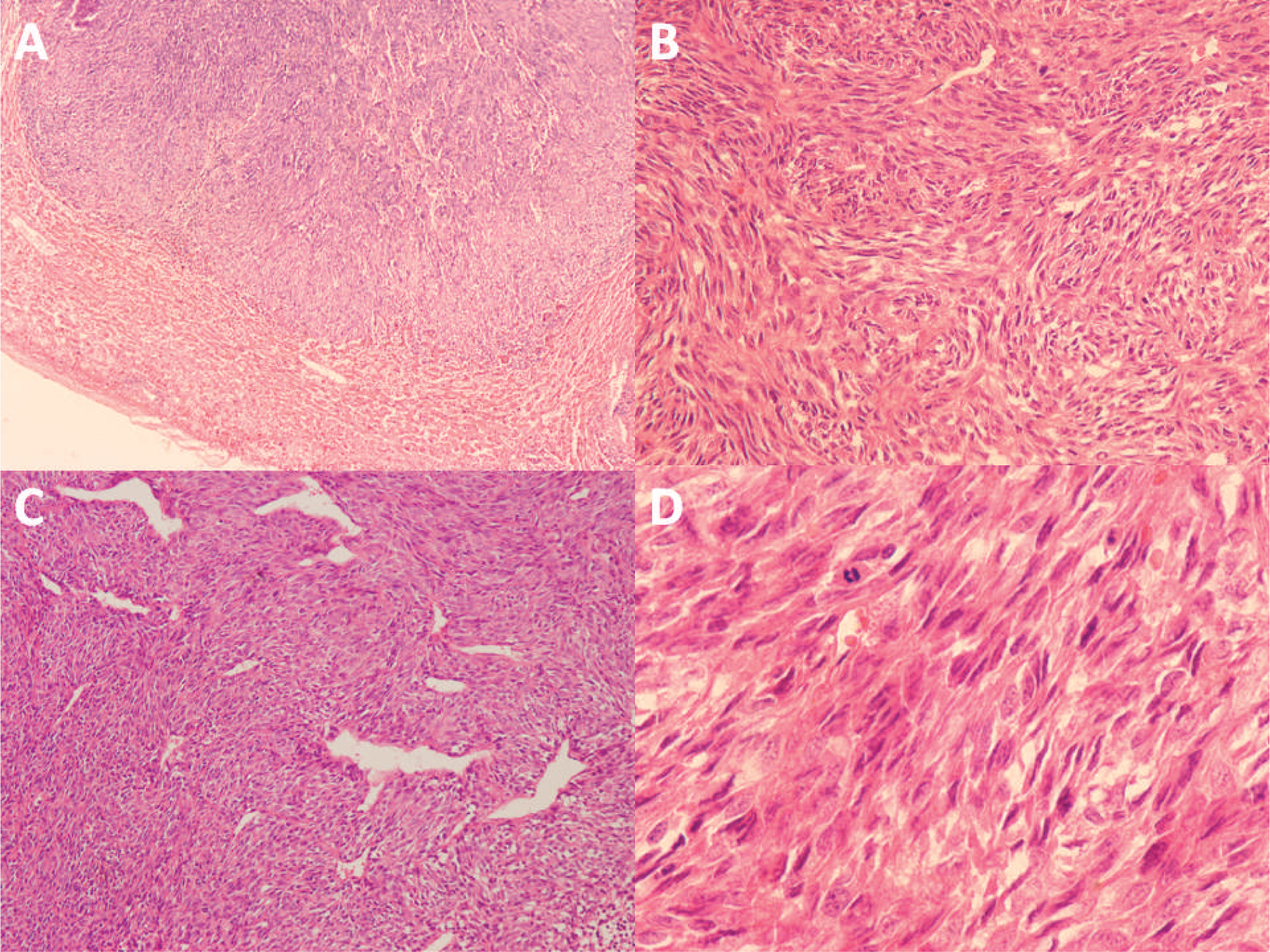
Histologic findings of the resected specimen.
Immunohistochemically, the neoplastic cells showed strong positivity for vimentin, bcl-2 (Fig. 3A), and TLE-1 (Fig. 3B). Focal staining for epithelial membrane antigen (Fig. 3C) and CK7 was observed. The tumor cells were negative for DOG1, CD117, CD34, S-100 protein, desmin, smooth muscle actin, CD99, hepatocyte antigen, and HMB-45. The MIB-1 index was 10%. Fluorescence in situ hybridization showed the rearrangement of SS18 in 85% of the neoplastic cells (Fig. 3D). Reverse transcription polymerase chain reaction and DNA sequencing confirmed the presence of SS18-SSX1 in the neoplasm. Based on the aforementioned findings, the final diagnosis was a monophasic synovial sarcoma of the liver.
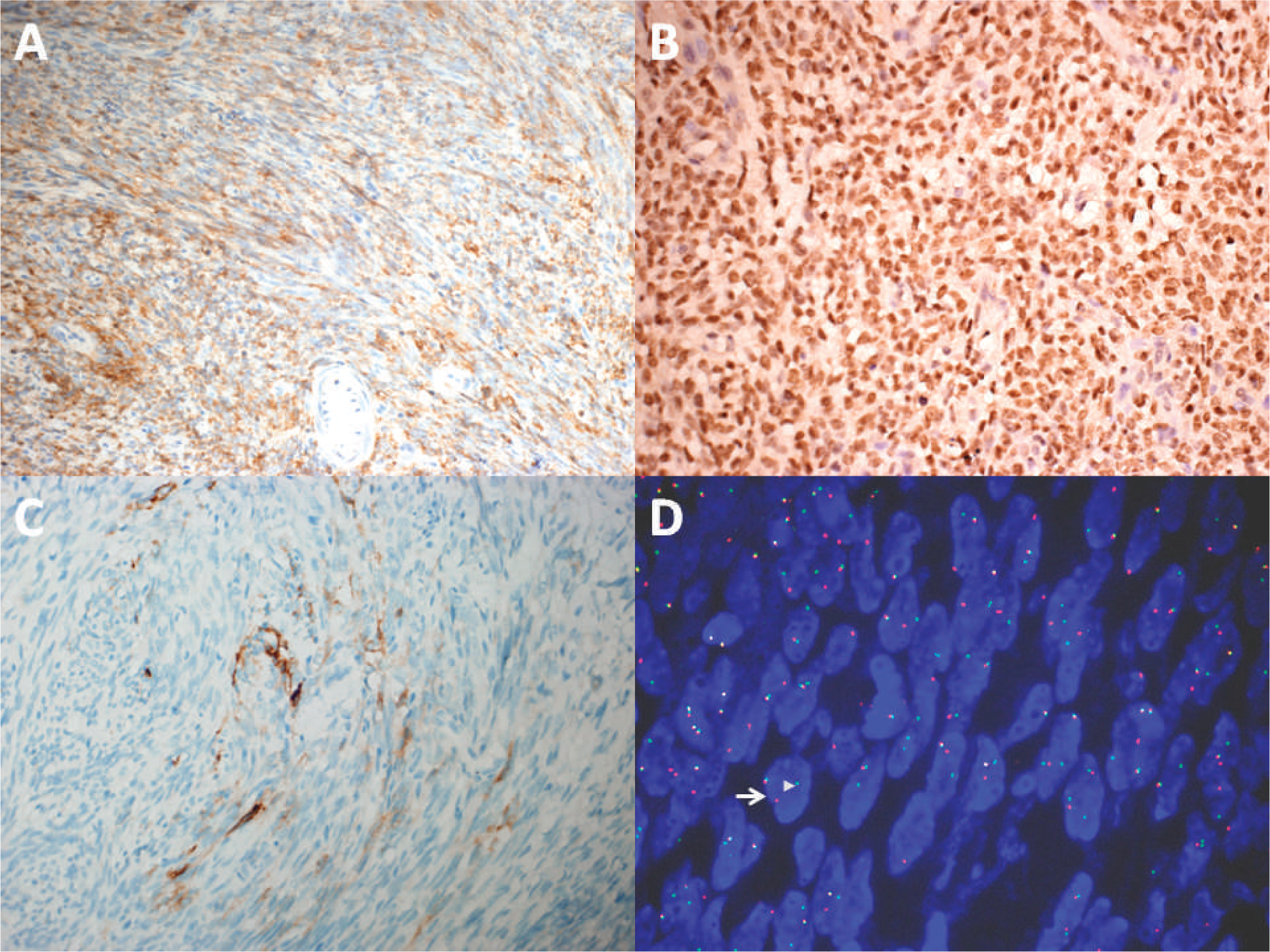
A detailed physical examination and positron emission tomographic scan did not show evidence of a tumor in other sites, especially the extremities. Symptoms were almost relieved completely, and the patient received ifosamide and adriamycin for 4 complete courses of chemotherapy in our hospital. Unfortunately, a new hepatic mass measuring 2.1 × 1.5 cm was detected at the site of operation by both ultrasonography and computed tomography in October 2012 (11 months after the surgery). However, the boy's parents did not accept any further therapy at this time, despite a recommendation. At the most recent follow up 16 months after the operation, he was still alive with relapsing tumor, and no evidence of metastasis was identified.
DISCUSSION
Synovial sarcoma is a morphologically, clinically, and genetically well-defined soft-tissue neoplasm, and most tumors occur in the extremities near the joints, followed by the head and neck region [1,2]. However, primary synovial sarcomas have also been reported to arise from a variety of unusual locations, such as mediastinum, retroperitoneum, and various viscera [3–5]. To the best of our knowledge, thus far only 3 cases of primary synovial sarcoma of the liver have been documented in the English literature [6–8]. The age of the 3 reported cases was 18, 44, and 60 years, respectively. Importantly, the current patient is the youngest one and represents the 1st example in the pediatric population. Microscopically, all of the 4 cases (including the current one) exhibited classic features of monophasic synovial sarcoma. Notably, the identification of exact fusion transcript (SS18-SS ×1) was done only in the present case, although all of the reported lesions were confirmed by cytogenetic or molecular genetic evidence of translocation t(X,18).
The differential diagnosis of primary synovial sarcoma of the liver is broad and might be very challenging, especially in small biopsy samples. This neoplasm should be differentiated from secondary synovial sarcoma and other more common types of primary pediatric liver neoplasms, such as nested stromal epithelial tumor (NSET), mesenchymal hepatoblastoma, and embryonal sarcoma. The differential diagnosis should also include other types of metastatic tumors.
Synovial sarcoma usually occurs in the extremities, and the liver is an extraordinarily rare location for this tumor type. Therefore, the possibility of a secondary synovial sarcoma should always be excluded, especially in the pediatric population. In the present case, both imaging studies and physical examinations ruled out the possibility of metastatic lesion.
NSET occurs predominantly in children and young adults. Additionally, this lesion is characterized by nests of spindle and epithelial cells, as well as calcifications or ossifications, simulating synovial sarcoma. However, NSET can be distinguished from synovial sarcoma in several aspects. First, NSET usually has an organoid appearance and lacks the diffuse fascicular pattern or hemangiopericytoma-like vascular pattern of synovial sarcoma [9]. Second, the majority of NSETs exhibit diffuse nuclear staining for WT-1, whereas most synovial sarcomas are negative for this antibody [10]. Third, and most important, identification of SS18-SSX fusion can be invaluable in distinguishing synovial sarcoma from NSET.
Mesenchymal hepatoblastoma should also be considered in the differential diagnosis whenever a spindle cell lesion of the liver is encountered in pediatric patients. The distinction can be aided through extensive sampling of the lesion to identify components of fetal or embryonal hepatocytes [9].
Embryonal sarcoma accounts for 13% of all hepatic neoplasms and usually develops in pediatric patients [11]. Sometimes the histologic features of embryonal sarcoma and poorly differentiated synovial sarcoma can overlap significantly. Importantly, a full panel of markers and molecular genetic testing can be useful to secure the diagnosis.
Hepatic synovial sarcoma can also mimic other types of metastatic neoplasms, such as gastrointestinal stromal tumor, desmoplastic small round cell tumor, solitary fibrous tumor, inflammatory myofibroblastic tumor, malignant peripheral nerve sheath tumor, Wilms tumor, and rhabdomyosarcoma. It is crucial for pathologists to keep in mind that a combination of clinical findings, careful morphologic examination, and ancillary studies, especially molecular studies, is helpful in distinguishing these lesions.
It is also noteworthy to mention that carcinoma is the most common tumor type in this site and can occur in children [11]. However, sarcomatoid carcinoma usually occurs in elder patients and is extremely rare in children.
In summary, primary hepatic synovial sarcoma is an extremely rare neoplasm that may occur in children, and therefore it should be rigorously distinguished from other spindle cell neoplasms.