
Abstract
Recurrent genetic alterations found in hepatic mesenchymal hamartoma include either androgenetic-biparental mosaicism or chromosomal rearrangements involving chromosome 19q13.4, in the vicinity of the chromosome 19q microRNA cluster (C19MC). Abnormal activation of C19MC, which is subject to paternal imprinting and is normally expressed only in placenta, could account for both genetic associations because androgenetic cells carry only paternal chromosomes. In this study, a 4.2-Mb deletion involving the 5′-end of C19MC was detected in a sporadic mesenchymal hamartoma by chromosomal microarray. Fluorescence in situ hybridization studies showed that the deletion localized to mesenchymal cells in the stroma of the hamartoma. Quantitative real-time polymerase chain reaction analysis of this tumor, 9 other sporadic hepatic mesenchymal hamartomas, and 3 hamartomas associated with androgenetic-biparental mosaicism demonstrated C19MC microRNA expression in all but 2 sporadic cases, with no significant expression in control liver. The findings support a pathogenetic model for mesenchymal hamartoma as a consequence of “ectopic” activation of C19MC in hepatic stroma, due to either chromosomal rearrangements or paternal uniparental disomy.
INTRODUCTION
Hepatic mesenchymal hamartoma (HMH) is a benign tumor comprising disorganized islands of hepatic epithelial cells, spaces lined by bile duct epithelium, and abundant, often myxedematous stroma that can undergo cystic degeneration. Although rare adult examples have been described, the overwhelming majority of cases occur in children, particularly in the first 2 years of life. Hepatic mesenchymal hamartoma is usually encountered as a sporadic lesion, but associations with Beckwith-Wiedemann syndrome and placental mesenchymal dysplasia are well documented [1–9].
Involvement of 1 or more parentally imprinted genetic loci in the pathogenesis of HMH has been speculated to account for association with Beckwith-Wiedemann syndrome and placental mesenchymal dysplasia [10–15]. Most cases of placental mesenchymal dysplasia and some examples of Beckwith-Wiedemann syndrome appear to be caused by androgenetic-biparental mosaicism (ABM), a condition in which androgenetic cells in the placenta and/or fetus contain only paternal chromosomes. Maternal imprinting is likely lost at all loci in androgenetic cells. We previously identified ABM in 2 cases of HMH, one with no history of placental mesenchymal dysplasia, and showed that the androgenetic cells are enriched in the stroma of the tumors [6,16]. However, molecular genetic studies of 9 other sporadic HMH cases failed to identify ABM.
The frequent documentation of chromosomal rearrangements involving chromosome 19q13.4 in sporadic HMH suggests involvement of 1 or more genes at this specific locus [17–30]. The most frequently reported rearrangements are translocations between chromosomes 11q and 19q13.4, one of which was characterized and shown to involve the MALAT1 gene at 11q13 and a site at 19q13.4, which initially did not contain an identifiable gene [26]. However, in the latter case, as well as another 3 pediatric HMHs analyzed by next-generation sequencing, the 19q13.4 breakpoints localize to a CpG-rich region that functions as a differentially methylated regulatory element for the chromosome 19q microRNA cluster (C19MC) [30].
C19MC is a tandem arrangement of 46 primate-specific microRNAs (miRNAs), which are each located within the introns of a single 37-exon RNA transcript [31]. Normal expression of the C19MC gene (C19MC) is restricted to the placenta, where differential methylation of the aforementioned CpG region correlates with selective expression of the paternal allele (parental imprinting) [32]. C19MC miRNAs are not expressed in liver or other normal nonplacental tissues, but transcriptional activation has been observed in a variety of neoplasms (eg, thyroid adenoma, primitive neuroectodermal tumors of the central nervous system, hepatocellular carcinoma) [33,34]. We hypothesized that activation of C19MC plays an important pathogenetic role in the sporadic and ABM-associated HMH.
METHODS
Cases and controls
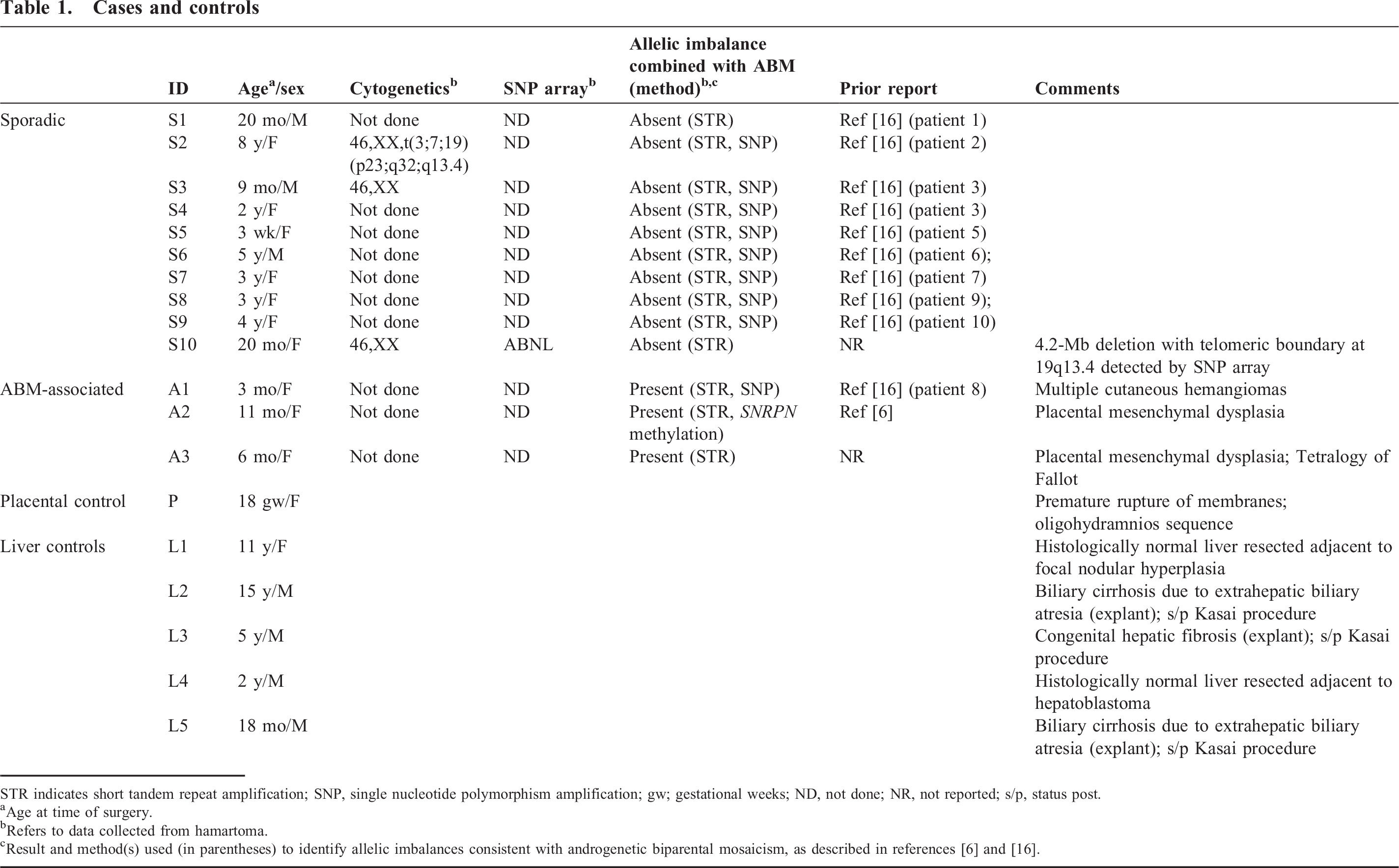
The investigation was conducted with approval from the Institutional Review Board at Seattle Children's Hospital. A search for archival specimens yielded paraffin blocks and hematoxylin and eosin (H&E)–stained slides from 13 hepatic resections for mesenchymal hamartoma, as well as 5 control hepatic resections or explants for nonhamartomatous disease and a full-term placenta with no diagnostic alterations. The slides were reviewed to select blocks composed primarily of hamartoma or adjacent histologically uninvolved liver. In some instances, original blocks were melted and the tissues trimmed and re-embedded to create new blocks composed as completely as possible of either hamartoma or uninvolved liver. For RNA extractions, 6–8 sections, each 10 μm thick, were collected. Available clinical records were reviewed from each case, with particular attention to any indications of Beckwith-Wiedemann syndrome, placental mesenchymal dysplasia, multiple hemangiomas, or other risk factors for ABM. As discussed in the “Results” section, all but 2 of the HMH cases were part of prior investigations of ABM-associated HMH, which did not address C19MC activation [6,16].
Detection of allelic imbalance by polymerase chain reaction (PCR) amplification of polymorphic short tandem repeats
DNA was extracted from frozen tumor (patient S10), paraffin sections (8 sections, 10 μm thick) of hamartoma and liver (patient A3), and white blood cells (parents of patient A3). Short tandem repeat (STR) loci, each located on a different chromosome, were investigated using the AmpFlSTR Profiler plus PCR amplification kit (Applied Biosystems, Grand Island, NY, USA), according to the manufacturer's instructions. The kit contains reagents to co-amplify 10 STRs: D3S1358, vWA, FGA, D8S1179, D21S11, D18S51, D5S818, D13S317, D7S820, and AMELOGENIN. Polymerase chain reaction products were separated on an ABI310 genetic analyzer (Applied Biosystems) with ROX internal lane standard. Amplitudes of the electropherogram peaks for each informative STR pair were measured, and the relative peak height (“peak ratio”) was used to detect allelic imbalance. To minimize potential bias introduced by more efficient amplification of shorter than of longer STRs, peak ratios were always expressed as the amplitude of the shorter STR divided by the amplitude of the longer allele. In addition, any locus with very poor PCR amplification (one or both peak heights less than 100 RFU) was discarded.
Chromosomal microarray
DNA extracted from cultured tumor cells and frozen tumor tissue was processed and hybridized to Affymetrix Cytoscan HD arrays (Affymetrix, Santa Clara, CA, USA), which contain both copy number and single nucleotide polymorphism probes, according to the manufacturer's protocols. Analysis was performed and results were visualized using ChAS v 1.2.2 software (Affymetrix).
Cytogenetics and fluorescence in situ hybridization (FISH)
Using a previously described procedure [35], cells from collagenase-disaggregated tumor (patient S10) were cultured in the referenced medium with AmnioMAX C-100 Supplement (Life Technologies, Carlsbad, CA, USA) added and were harvested after 7 days to yield a cell pellet of nuclei in Carnoy's fixative (3∶1 methanol∶acetic acid). From this pellet, slides were prepared and G-banded with Wright's stain for karyotype analysis.
For FISH, a BAC probe within the region of deletion on the long arm of chromosome 19 that was defined by chromosomal microarray analysis (RP11-11OI11; Signature Genomics Laboratories, LLC, Spokane, WA, USA) was labeled with an orange fluorophore using a nick translation kit (Spectrum Orange; Abbott Molecular Laboratories, Abbott Park, IL, USA) according to the manufacturer's instructions. As an internal control, this probe was mixed with a green fluorophore–labeled FISH probe from the subtelomeric region of the short arm of chromosome 19 (19pter; Cytocell Ltd, Cambridge, United Kingdom) to create a dual-color probe mixture. Interphase FISH using the dual-color probe mixture was performed on unstained slides prepared from the fixed pellet using conventional procedures. Briefly, the slides were pretreated 2×saline sodium chloride (SSC) (37°C; 30 minutes), dehydrated in a 70%, 80%, 100% ethanol series (4°C; 2 minutes each), air-dried, denatured in 70% formamide/2×SSC (72°C; 1 minute), and dehydrated in the ethanol series again. The dual-color probe was denatured (72°C; 5 minutes), applied to the slide, and incubated overnight at 37°C. The slides were then washed sequentially in 0.4×SSC/0.1% NP-40 (72°C; 2 minutes) and 2×SSC/0.1% NP-40 (RT; 2 minutes), and DAP II counterstain/antifade (Abbott Molecular Laboratories) was applied.
For interphase FISH to the paraffin-embedded tumor, 4-μm sections were placed on slides and deparaffinized using standard procedures. The slides were then treated sequentially as follows: 0.2 N HCl (RT; 20 minutes), sterile water (RT; 3 minutes), 2×SSC (RT; 3 minutes), 1 M sodium isothiocyanate (80°C; 20 minutes), sterile water (RT; 3 minutes), 2×SSC (RT; 5 minutes), 0.5 mg/mL pepsin in saline (pH 2; 37°C; 20 minutes), 2 washes of 2×SSC (RT; 5 minutes), denatured with the dual-color probe mixture (73°C; 5 minutes), and incubated overnight at 37°C. The slides were then washed in 2×SSC/0.3% NP-40 (73°C; 2.5 minutes), and DAP II counterstain/antifade was applied.
Slides were evaluated by standard epifluorescence microscopy, and images were captured using CytoVision software (Applied Imaging Corp, Santa Clara, CA, USA).
Quantitative reverse transcriptase (RT)–PCR of C19MC miRNAs
Total RNA was isolated from frozen samples of patient S10′s hamartoma, a cell culture of this hamartoma, normal control liver, and normal, control term placenta, as well as from paraffin sections from all of the hamartomas in the series, adjacent histologically normal liver, control nonhamartomatous livers, and normal, control term placenta. For both frozen and paraffin-embedded tissues the RecoverAll Total Nucleic Acid Isolation Kit (Life Technologies, Grand Island, NY, USA) was used, according to the manufacturer's instructions, and RNA was quantified spectrophotometrically. Quantitative RT-PCR was performed to target a subset of 4 C19MC miRNAs (miR-518d, miR-520c, miR-526a-1, and miR-526-2) and RNU6B, with the TaqMan MicroRNA Assay (Catalog No. 4427975, Life Technologies), according to the manufacturer's instructions. All samples were run in triplicate along with in parallel with control liver and placenta RNA extracts. Mean values were obtained and relative quantification (RQ) was calculated using Applied Biosystems SDS software based on the RQ = 2_DDCt 2(-DeltaDelta C(T)) method [36]. Ct data were normalized to an internal small RNA control, RNU6B, as described elsewhere [37]. Normal placenta and liver were run with every set of samples, and the mean value obtained from placenta was used as a reference against which to express relative levels of miRNA from other samples. After 40 cycles, a 1-μL aliquot of representative products from each sample was analyzed electrophoretically using the Agilent 2100 Bioanalyzer (Agilent Technologies, Wilmington, DE, USA), according to the manufacturer's instructions.
RESULTS
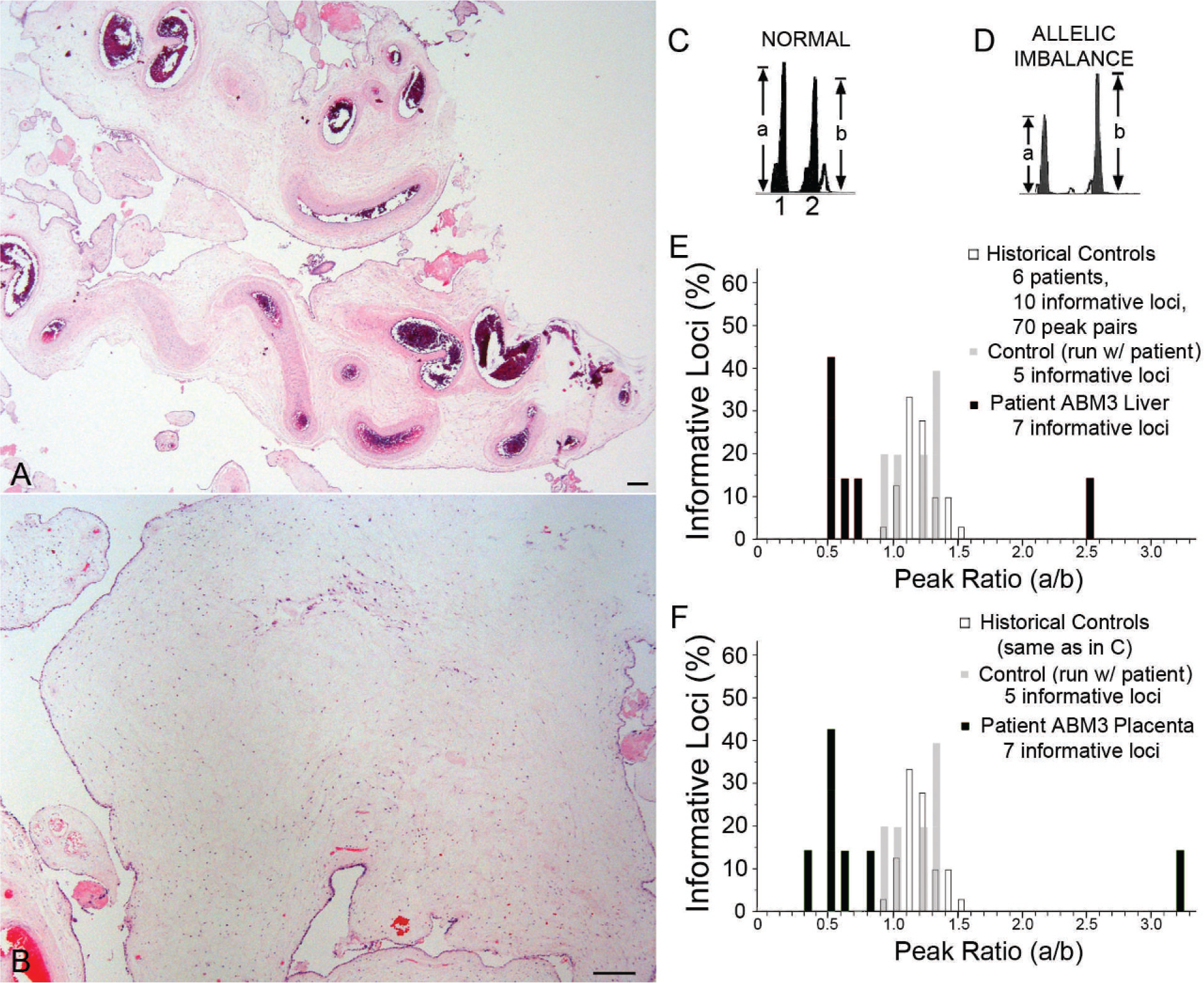
The clinical data available for the series of HMHs analyzed in this study are shown in Table 1. Most of these tumors were tested for ABM by a variety of multiple molecular genetic approaches, and the results were published previously [16]. The subset of 3 ABM-associated HMHs includes the tumors reported by Reed and colleagues [6] and Lin and colleagues [16], as well as a previously unreported case (patient A3). At the time the latter hamartoma was resected, the patient was a 6-month-old girl, who was born at 29 weeks' gestation with Tetralogy of Fallot, liver cysts, and a cystic placenta. The partially disrupted placenta weighed 806 g and measured 23 × 19 × 4 cm. The fetal surface had a 7 × 4.5–cm area of subamniotic, hyperemic grittiness (30% of the identifiable fetal surface). Beneath this region and in multiple other foci, the villi were markedly cystic and filled with a clear, tan, viscous fluid (approximately 30% of the specimen). Noncystic placental parenchyma was spongy, soft, and tan-red, but with markedly tortuous vasculature. Microscopic examination of the placenta revealed a subset of enlarged hydropic villi, including stromal cystic degeneration (Fig. 1A,B), without trophoblast proliferation; other intermixed villi were small and appropriately sized for an estimated gestational age of 29–30 weeks. Many large, tortuous vessels, some with fibrous intimal hyperplasia, were present in edematous stem villi.
Placental mesenchymal dysplasia (
Cases and controls
STR indicates short tandem repeat amplification; SNP, single nucleotide polymorphism amplification; gw; gestational weeks; ND, not done; NR, not reported; s/p, status post.
Age at time of surgery.
Refers to data collected from hamartoma.
Quantitative analysis of PCR products from a battery of polymorphic STR elements yielded allelic imbalances consistent with ABM (7 informative loci), with an overrepresentation of paternal alleles at the 4 loci for which parental alleles could be distinguished. Allelic imbalance was detected in DNA extracted from the hepatic hamartoma and placenta, but not the infant's blood (Fig. 1C–F). Flow cytometric DNA ploidy studies of the placenta excluded triploidy.
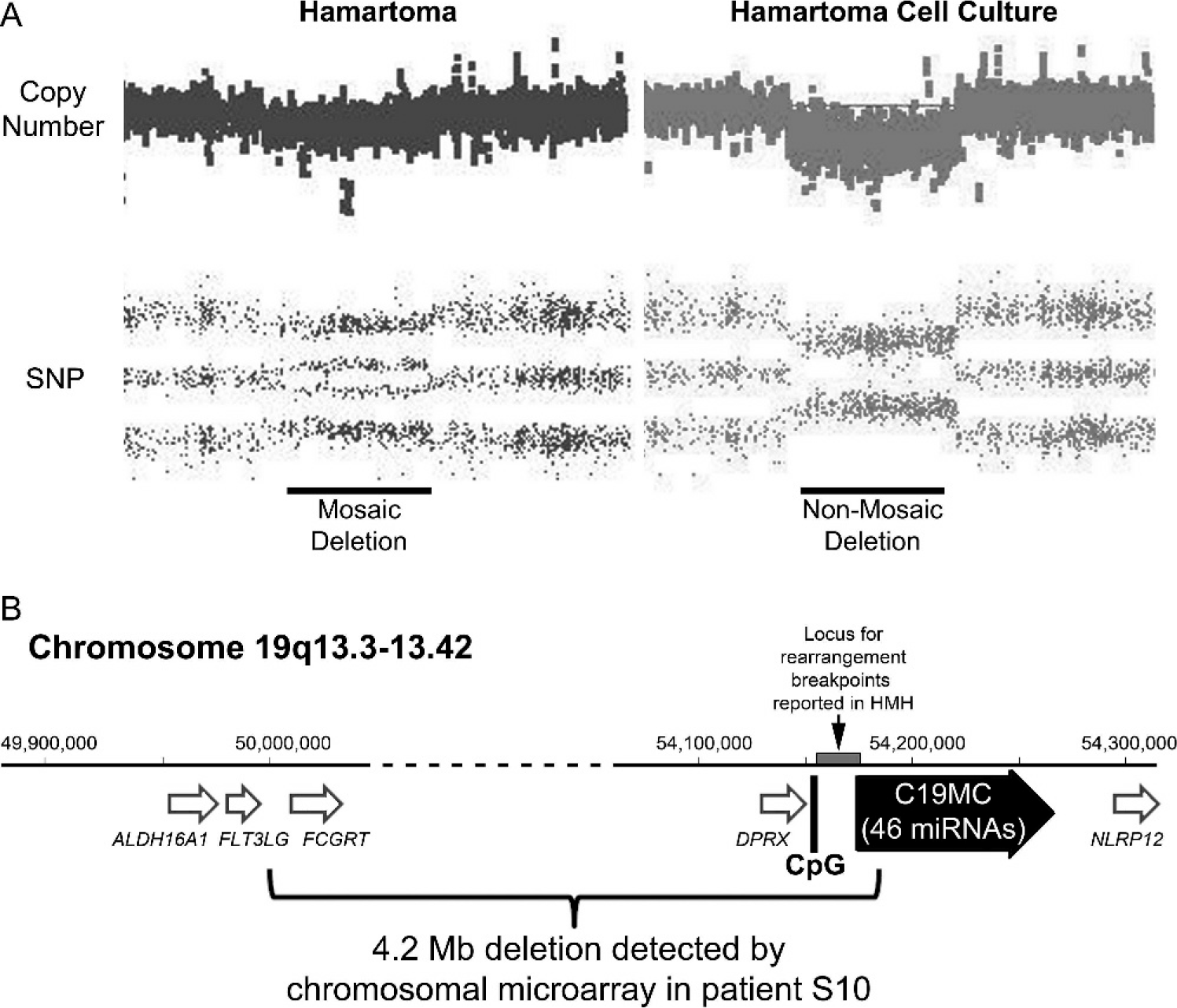
Among the 10 examples of sporadic HMH in the series, no molecular evidence of ABM was detected in 9, as reported elsewhere [16]. The remaining patient (S10) presented at age 20 months after a well-child check revealed a palpable abdominal mass. She was the product of a naturally conceived twin gestation, and her male co-twin reportedly died as a neonate as a result of renal agenesis. No information was available regarding examination of the placenta postdelivery, but according to the mother, prenatal ultrasound examinations did not disclose any obvious anomalies with either twin's placenta. Abdominal imaging of the infant demonstrated a large heterogeneous, partially cystic mass in the inferior right lobe of the liver, which was resected. The 657-g mass had typical gross and microscopic features of hepatic mesenchymal hamartoma (Fig. 2). Cytogenetic studies of twenty G-banded metaphase cells cultured from the mass yielded an apparently normal karyotype (46,XX), but chromosomal microarray analysis of the DNA extracted directly from the noncultured hamartoma demonstrated nonmosaic loss of heterozygosity, indicative of a 4.2-Mb deletion of chromosome 19q (Fig. 3A). Similar microarray studies of cells cultured from the hamartoma, which morphologically resembled fibroblasts in vitro, were consistent with a nonmosaic cell population carrying the same deletion (Fig. 3A). The presence of mosaicism for the deletion in DNA extracts directly from the tumor, which also contained nonneoplastic cells, confirmed the acquired (nonconstitutive) nature of the deletion. The boundaries between intact and deleted probes coincided with a centromeric deletion boundary in intron 8 of the FLT3LG gene and a telomeric deletion boundary in intron 5 of C19MC (Fig. 3B). Multiple efforts to clone the breakpoint using various PCR-based strategies were unsuccessful, and a Southern blot analysis suggested a more complex rearrangement than a simple interstitial deletion (data not shown).
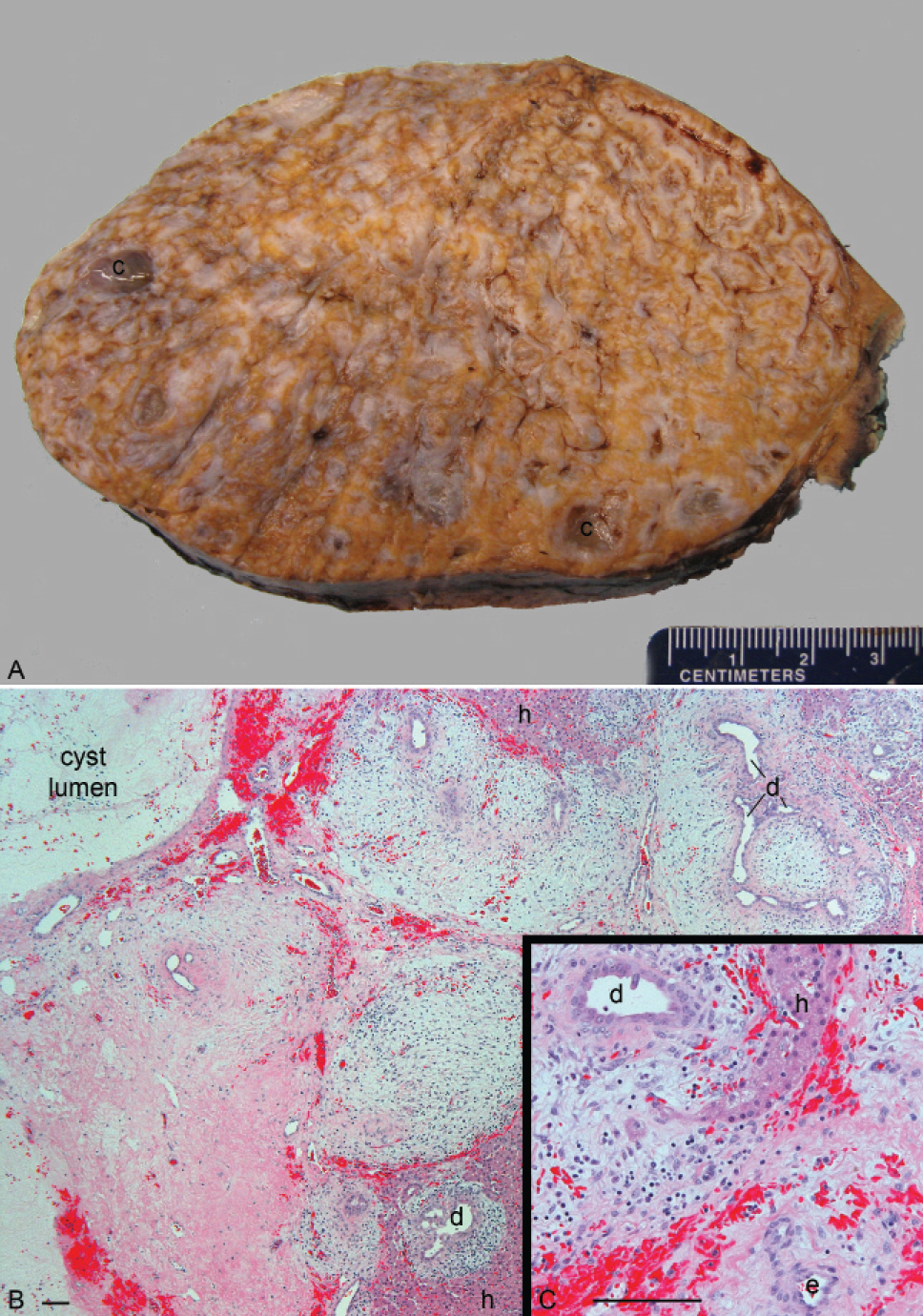
Gross and microscopic images from patient S10′s hamartoma. (
(
Fluorescence in situ hybridizaton with a probe specific for the deleted region yielded results expected from the chromosomal array data. The overwhelming majority (43/50, 86%) of cells cultured from the tumor had only one 19q signal (Fig. 4), versus 4/100 (4%) in a diploid cell control, suggesting that the deletion might be prevalent in stromal cells. This was confirmed by interphase FISH on paraffin sections from the tumor, which demonstrated significantly more cells with 0 or 1 19q hybridization signal in tumor stromal cells, as compared with stromal cells in adjacent histologically normal liver, epithelial cells in the tumor, or epithelial cells in adjacent liver (Fig. 4).
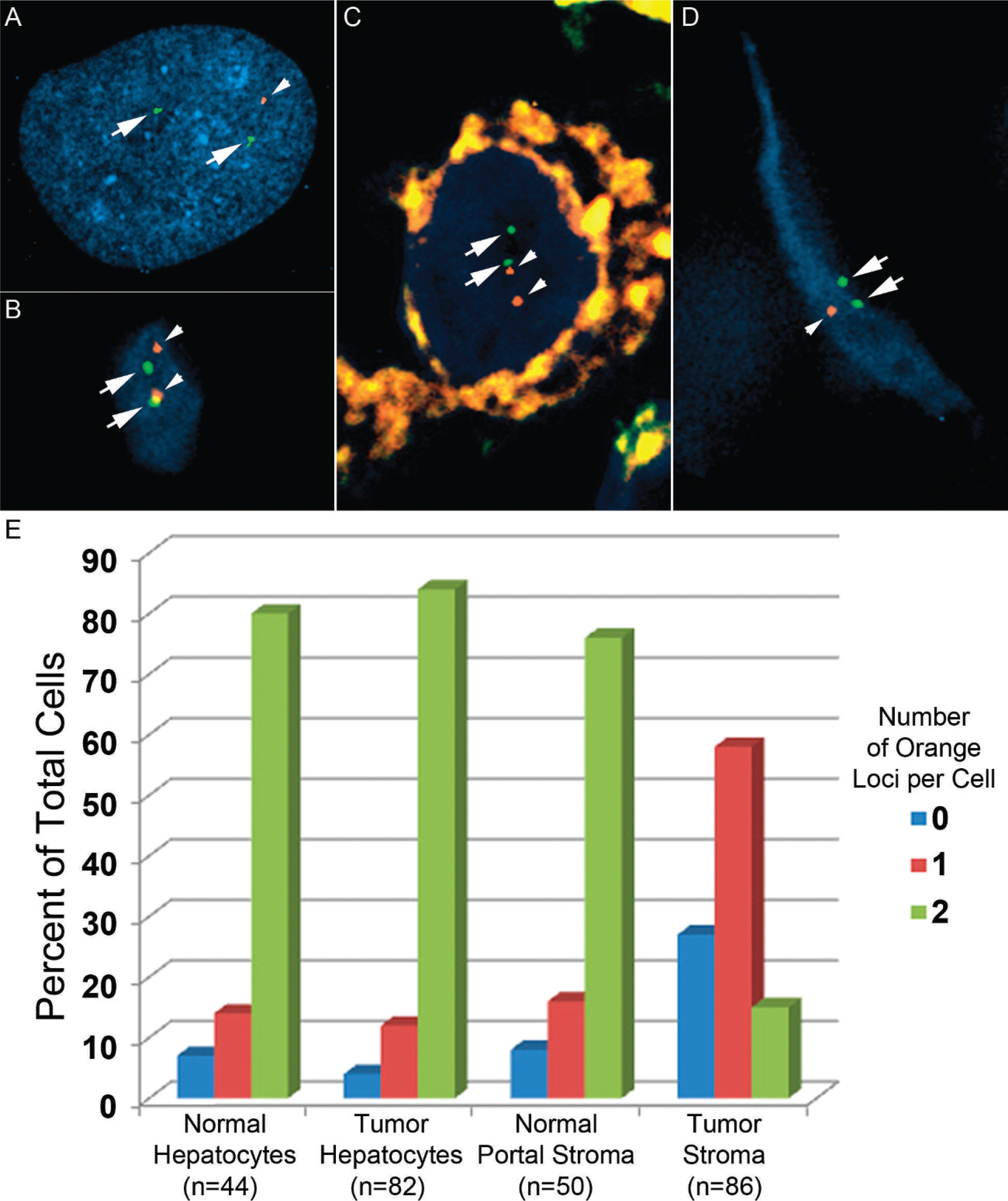
Interphase fluorescence in situ hybridization (FISH) demonstrates overrepresentation of cells with 19q13.4 deletion in mesenchymal cells of hepatic mesenchymal hamartoma; FISH was performed on cells grown in vitro (
Like the chromosomal rearrangements reported in other sporadic HMH, the deletion in our patient's tumor disrupts the imprinted gene, C19MC. We hypothesized that “ectopic” activation of some or all miRNAs encoded by C19MC might have resulted from these rearrangements in sporadic HMH or abnormal imprinting in ABM-associated HMH. For all 13 hamartomas in this series, C19MC miRNA expression was evaluated by quantitative RT PCR. Levels of expression were normalized against RNU6B, a constitutively expressed small nuclear RNA, and compared with the levels of C19MC miRNA in normal placenta, control liver with no history of hamartomatous liver disease, and adjacent nonhamartomatous liver from the same patient, when available. In general, no significant C19MC miRNA (less than 1000 times the amount present in placenta) was detected in nonhamartomatous liver controls, including adjacent nonhamartomatous liver from 10/12 cases (Fig. 5A). In contrast, abundant C19MC miRNA was present in all but 2 hamartoma samples. Excluding the latter 2 cases (S1 and S5), the relative levels of miRNA detected in the hamartomas ranged from 10% to 110% of those observed in control placenta and were 103 to 104 times greater than in normal liver. Gel electrophoresis of the PCR products confirmed specific amplification of the expected 52-bp C19MC miRNA product (Fig. 5B) in the samples with high levels of miRNA detected by quantitative RT-PCR.
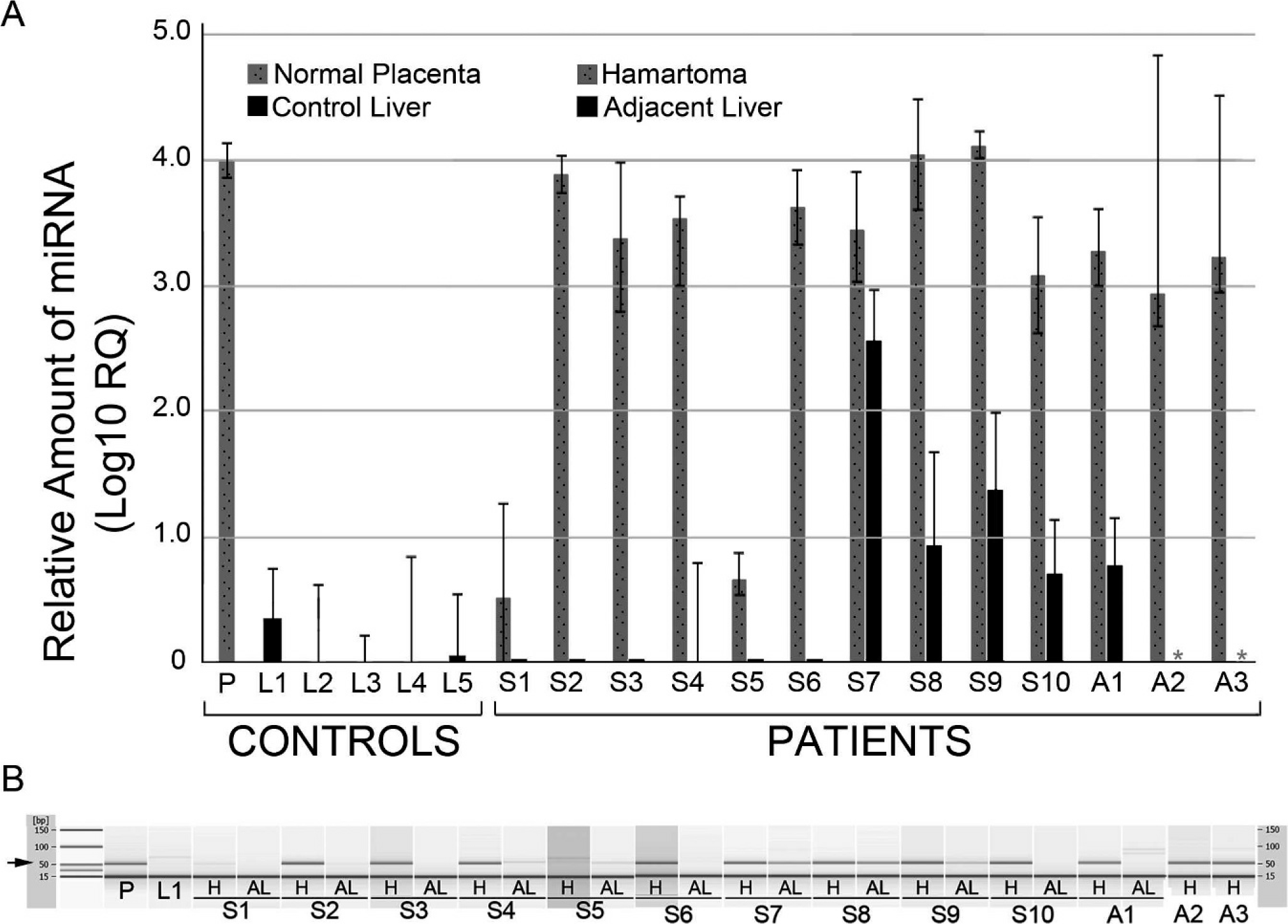
(
DISCUSSION
The results of this investigation support a pathogenetic model of HMH that involves C19MC activation in at least 2 genetic contexts. The 1st is chromosomal rearrangements that disrupt C19MC and are frequently observed in sporadic HMH. The 2nd is disregulation of C19MC, likely caused by paternal uniparental disomy in the androgenetic cells of patients with ABM.
The interstitial deletion reported in patient S10 adds to a list of over 15 reported examples of sporadic HMH with 19q13.3–q13.4 rearrangements, the locus in which C19MC resides (see table 2 in Mathews and colleagues [30]). The disruptions in 4 of these cases have been characterized further. From one hamartoma, Rajaram and colleagues [26] cloned the breakpoints of an 11;19 translocation and reported DNA sequences located between the differentially methylated CpG island that regulates C19MC expression and the 1st exon. Mathews and colleagues [30] used next-generation sequencing to evaluate 3 other sporadic pediatric HMHs and found disruptions in the same locus (Fig. 3), including an inversion with a breakpoint located 3′ to the 1st exon. Our microarray data appear to expand the spectrum of disruptions within this locus to include a case in which the first 5 exons and upstream CpG-rich promoter appear to be deleted. The C19MC expression data presented here provide the first direct evidence that activation of the miRNA cluster may be an important consequence of these rearrangements.
The precise mechanism by which C19MC was activated in patient 10 or any other reported example of sporadic HMH remains to be determined. It seems likely that translocations, deletions, or other structural rearrangements juxtapose heterologous regulatory regions for other genes with exons and introns of C19MC, causing misexpression of the latter in the liver. In those cases with 11;19 translocations, DNA analysis suggests that the MALAT1 gene is partnered with C19MC, whereas in our patient with an interstitial deletion, a potential fusion transcript involving FLT3LG and the terminal 32 exons of C19MC should be considered. To date, preliminary efforts in our laboratory to capture a putative fusion product using RT-PCR have been unsuccessful (data not shown), but a variety of strategies might be employed to continue this search.
Stromal hyperplasia is a histopathological hallmark of HMH, but mature epithelial components are invariably present as well. The results from FISH of paraffin sections and cultured cells from the hamartoma with an interstitial chromosome 19q deletion strongly suggest that it is the fibroblast-like cells in the stroma that harbor the genetic alteration that likely “drives” hamartoma formation. Mathews and colleagues [30] observed a similar stroma-specific localization of chromosome 19q rearrangements in their series of 3 pediatric HMHs. Thus, bile ducts, islands of hepatocytes, and often massive cysts lined by bile duct epithelium are prevalent in HMH but likely represent deranged liver development brought about by clonal genetic alterations in hepatic mesenchymal tissue.
In our series, activation of C19MC was a shared feature of most sporadic (8/10) and ABM-associated (3/3) HMHs. In 2 sporadic tumors (S1 and S5), no significant expression of C19MC miRNAs was detected. This may reflect true heterogeneity in HMH, whereby lack of C19MC activation is not a feature in an apparent small minority of cases. However, as our assay only evaluated 4 of the 46 miRNAs encoded by the full-length C19MC transcript, activation of one or more other C19MC miRNA cannot be excluded. Indeed, while the 4.2-Mb deletion in patient S10 eliminates the first 5 exons of the gene, this rearrangement appears to promote ectopic expression of miRNAs located in downstream introns, including those detected by quantitative RT-PCR in this investigation. Perhaps one or both of the hamartomas with no significant measurable miRNA in our series harbor 19q rearrangements that activate miRNAs located in introns even further downstream than those we evaluated.
With 1 exception (S7), no or minimal C19MC miRNA was detected in histologically nonhamartomatous liver adjacent to HMH. These results provide excellent internal controls for our HMH cases and are consistent with published data that indicate C19MC is only expressed normally in the placenta [32]. In this context, the relatively high level of miRNA detected in liver adjacent to HMH S7 is unexpected. Examination of H&E-stained sections from the same tissue block and flanking those used for RNA extraction only reveal histologically normal liver with intact lobular architecture and no significant fibrosis. It is possible that hamartomatous cells have infiltrated the tissue in a histologically inconspicuous manner. Regardless, the case is an outlier in comparison with the others. A 2nd case (S9) had low levels of miRNA (∼800× less than in placenta), but levels that were slightly above background levels. In this instance, H&E-stained sections showed inadvertent inclusion of a small portion of hamartomatous stroma at the edge of the sections used for RNA extraction.
Unfortunately, none of the 3 ABM-associated tumors was sampled for conventional cytogenetics or chromosomal microarray, so structural rearrangements of C19MC cannot be excluded. However, we suspect a different mechanism of C19MC activation underlies ABM-associated HMH for several reasons. At least 7 examples of HMH have been reported with placental mesenchymal dysplasia and/or ABM [4–9,16]. Given the rarity of HMH, this number of associations is higher than expected by chance and suggests a shared pathogenetic basis. Since C19MC is an imprinted gene, normally expressed only in placenta from the paternal allele, its regulation may be disrupted in androgenetic cells with 2 paternal alleles and no maternal allele. The basis for silence of C19MC in liver or other nonplacental sites is unknown, but for other imprinted genes with differentially methylated regulatory regions, trans interactions between maternal and paternal alleles are important, and loss of 1 parental allele leads to disregulation of the other [38]. Finally, microdissection studies of a case of ABM-associated HMH indicated that the androgenetic cells are enriched in the stroma, not the epithelium [16]. This distribution of androgenetic cells is analogous to the stromal localization of cells with C19MC rearrangements in sporadic HMH and suggests that pangenomic paternal uniparental disomy in hepatic stroma may have similar downstream molecular effects.
We speculate that paternal uniparental disomy in androgenetic hepatic stromal cells leads to abnormal activation of C19MC, thereby mimicking the molecular effect of HMH-associated structural rearrangements at the same locus. This model leads to logical hypotheses that we hope to address in future investigations, including the methylation status of the CpG island in ABM-associated HMH and downstream targets for C19MC miRNAs. Identification of pathogenetically significant miRNA targets will be a challenge because over 4500 putative mRNA substrates have been identified in human embryonic stem cells [39].
In summary, activation of the primate-specific C19MC appears to be a consistent feature of sporadic or ABM-associated HMH and probably functions within stromal cells to promote hamartoma formation.