
Abstract
We report the postmortem findings of two siblings with gross and microscopic features consistent with IMAGe association (Intrauterine growth retardation, Metaphyseal dysplasia, Adrenal hypoplasia congenita, and Genital anomalies) with an emphasis on the histopathology of the adrenal gland in this rare syndrome. The first sibling was an 8-week old male diagnosed postnatally with primary adrenal insufficiency. There was no deletion of the DAX1 gene by FISH. Examination at autopsy revealed dysmorphic features including frontal bossing, epicanthal folds, flat philtrum, cryptorchidism, penile chordee, overriding fourth toe, and height and weight below 3rd percentile. Grossly, the adrenal glands were not identified; however, microscopic examination of the suprarenal soft tissue revealed a 3 mm focus of disorganized fetal adrenal cortex with distended “cytomegalic” cells with abundant pink eosinophilic cytoplasm, vesicular nuclei, and cytoplasmic vacuolization. A minute focus of permanent adult cortex was also seen, but no adrenal medulla was identified. An autopsy of the sibling, who died 12 years previously at day 9 of life, revealed dysmorphic facial features with cryptorchidism and a large phallus. The adrenal glands were grossly hypoplastic (11 mm). Histologically, the adrenal glands showed disorganized fetal cortex with cytomegalic cells, a larger amount of permanent adult cortex, and bizarre nuclei with numerous pseudoinclusions. While there is currently limited information regarding the histopathologic adrenal findings in IMAGe association, our small case series suggests overlapping features between X-linked recessive congenital adrenal hypoplasia (cytomegalic cells with lack of permanent adult cortex) and autosomal recessive congenital adrenal hypoplasia (diminished permanent adult cortex without cytomegalic cells).
INTRODUCTION
Congenital adrenal hypoplasia is an uncommon, life-threatening condition with an estimated frequency of 1 in 12,500 live births. It often presents in infancy with symptoms of primary adrenal insufficiency and requires early detection with steroid replacement therapy for survival.
Two forms of congenital adrenal hypoplasia have well-documented adrenal histopathologic findings. These are the X-linked recessive and autosomal recessive forms of congenital adrenal hypoplasia (OMIM 300200). The X-linked recessive, “cytomegalic” form is associated with DAX1 (dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1) mutations on the X chromosome (Xp21.2) [1]. It has a male predominance and is associated with hypogonadotropic hypogonadism [2]. It can occur as an isolated abnormality or as part of a contiguous gene deletion at the same location (Xp21), resulting in congenital adrenal hypoplasia, glycerol kinase deficiency, and Duchenne muscular dystrophy [3]. Histopathologic examination of the adrenal glands reveals disorganized and hypoplastic tissue consisting of pale to eosinophilic, cytomegalic cells with frequent intranuclear inclusions and lack of permanent adult cortex [4].
The autosomal recessive, “miniature adult” form of congenital adrenal hypoplasia, affects both genders. It is associated with mutations or deletions in the gene that encodes SF1 (steroidogenic factor 1) on chromosome 9q33 [5]. Histopathologic examination of the adrenal glands reveals hypoplastic tissue lacking fetal cortex, consisting solely of permanent adult cortex without cytomegalic cells [6].
IMAGe association (OMIM 614732) is another cause of congenital adrenal hypoplasia that was first described by Vilain et al. in a case series involving three unrelated boys with intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital abnormalities [7]. IMAGe has been shown to be associated with heterozygous mutation in the CDKN1C gene on chromosome 11p15.4, thought to be inherited via maternal imprinting [8]. Only twenty-one cases have currently been described in the literature. One case series, from Tan et al., describes the postmortem adrenal gland morphology of a three-month old girl with IMAGe association [9]. Our report describes two additional cases of IMAGe association with an emphasis on postmortem adrenal histopathologic findings.
CLINICAL PRESENTATIONS
Case 1
An African American male infant was born to a 36 year old mother (G11 P 1-3-7-2) with a history of seven prior spontaneous abortions during the first trimester (gender and cause unknown). She has two healthy living children aged 6 (female) and 7 (male). Prenatal ultrasound was concerning for achondroplasia. At 38 weeks gestation a Cesarean section was performed due to intrauterine growth restriction and non-reassuring fetal heart tones. Apgar scores were 4 and 9 at one and five minutes, respectively. Birth weight and length were below the 3rd percentile, and head circumference was above than 97th percentile. A postnatal skeletal survey showed no abnormalities. Dysmorphic features including diffuse skin hyperpigmentation, and genital abnormalities including undescended left testicle and penile chordee were noted on physical examination. Upper and lower extremities were shortened. The karyotype was normal (46, XY) with no significant abnormalities detected.
Immediately after birth, the infant developed respiratory distress and poor feeding with hypoglycemia and was admitted to the neonatal intensive care unit. He was placed on continuous positive airway pressure. He developed progressive hyponatremia and hyperkalemia at one week of life. Blood glucose levels normalized. Cortisol was undetectable, aldosterone was low (3.1 ng/dL), and both renin (137.70 ng/mL/hr) and ACTH (6269 pg/mL) were elevated. Testosterone was low (30.0 ng/dL), but LH (5.7 mIU/mL) and FSH (1.9 mIU/mL) were within normal limits. Growth hormone (19.5 ng/mL) and TSH (25.070 mIU/L) were both elevated. 17-hydroxyprogesterone (19.0 ng/dL) was within normal limits and 17-hydroxypregnenolone (201.0 ng/dL) was mildly decreased. Fluorescence in situ hybridization (FISH) analysis did not show deletion of the DAX1 gene. He was diagnosed with primary adrenal insufficiency, attributed to IMAGe association, and treated with stress-dose hydrocortisone which was weaned to maintenance therapy once he became more clinically stable. He also had hypocalcemia with hypercalciuria, which was treated adequately with calcium carbonate. He was discharged home on maintenance hydrocortisone, fludrocortisone, and both calcium carbonate and sodium chloride supplementation.
At eight weeks of life, he became limp and unresponsive necessitating cardiopulmonary resuscitation. There were no symptoms of acute illness prior to this event. On arrival at the emergency department, he was intubated and given methylprednisolone. Asystole persisted for greater than 20 minutes. Neurologic exam showed evidence of severe hypoxic-ischemic brain injury and support was withdrawn.
Case 2
Twelve years prior, the mother delivered a male infant at 33 weeks gestation via Cesarean section. Prenatal screening revealed elevated maternal serum alpha fetoprotein (AFP). Amniocentesis fluid AFP was normal. Karyotype was normal male (46, XY). Cesarean section was performed due to partial placental abruption and maternal pre-eclampsia with suspected intrauterine growth restriction. Apgar scores were 3 and 7 at one and five minutes, respectively. Birth weight was 1086 grams (below the 3rd percentile). Physical examination was notable for macrocephaly and genital anomalies including a large phallus and bilateral undescended testes. A postnatal skeletal survey was not performed at that time. His newborn course was complicated by thrombocytopenia, hyperbilirubinemia, neutropenia, occasional hyponatremia, acidosis, and acute respiratory distress. Empirical antibiotic therapy was initiated for culture negative sepsis and neutropenia was treated with granulocyte colony stimulating factor (G-CSF). TORCH screen was negative. Despite therapy, at nine days of life the infant developed apnea, hypotension and ultimately bradycardia and died.
PATHOLOGIC FINDINGS
Case 1
External examination revealed dysmorphic facial features including prominent frontal bossing, bilateral epicanthal folds, and flat philtrum. Other notable findings included widened anterior fontanelle, umbilical hernia, left-sided cryptorchidism, penile chordee, and overriding left fourth toe.
Examination of the central nervous system revealed diffuse hypoxic-ischemic changes involving the cerebral cortex, thalamus, hippocampus, brainstem, cranial nerve nuclei, cerebellum, and spinal cord, along with mild reactive gliosis of the white matter. The lungs had diffuse alveolar damage with hyaline membranes and vascular congestion. The heart had no abnormalities, including no evidence of acute myocardial infarction. The testes were histologically unremarkable.
The adrenal glands were not grossly identified. Microscopic examination of all suprarenal soft tissue revealed a single 3 mm focus of left-sided adrenal tissue. The tissue consisted of a nodular proliferation of disorganized fetal cortex with large, distended, eosinophilic and vacuolated cells with enlarged nuclei (adrenal cytomegaly), surrounded by paraganglia (Figure 1). A minute focus of apparent permanent cortex was also present, confirmed by immunoreactivity with Melan-A and inhibin (fetal cortex was also immunoreactive with Melan-A and inhibin but paraganglia were negative). No adrenal medulla was identified.
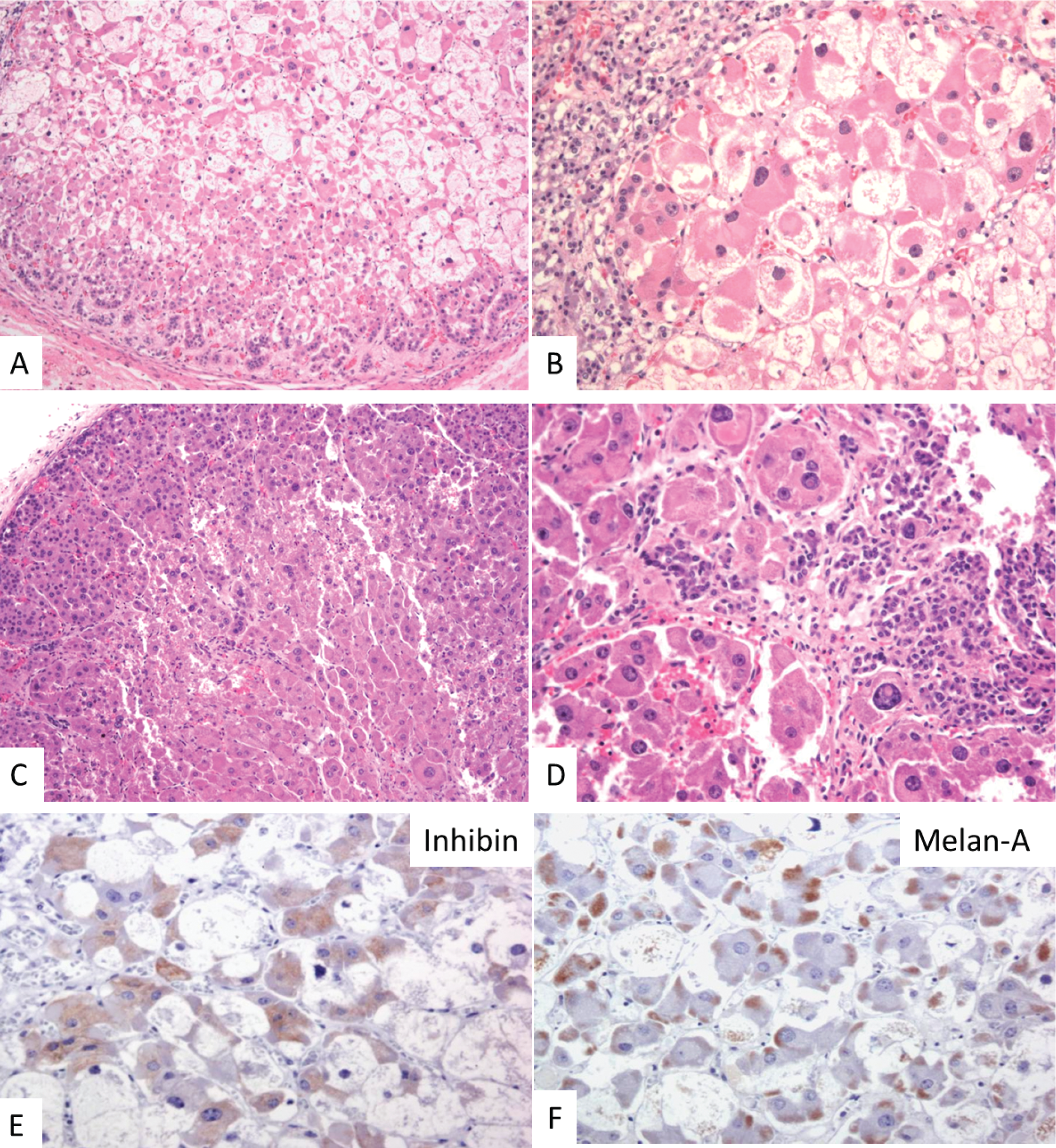
Adrenal gland histology in two cases of IMAGe association. (
Case 2
External examination revealed dysmorphic features including relative macrocephaly and genital anomalies including large phallus and bilateral cryptorchidism.
Examination of the central nervous system revealed diffuse congestion with a 0.5 cm organizing subacute subarachnoid hemorrhage overlying the left anterior cerebellar pole. The lungs were mildly congested with focal alveolar hemorrhage.
The adrenal glands were small and spherical, together measuring 11 mm and weighing less than 0.5 grams (normal range 5.1 g +/− 2.2 g). Microscopic examination revealed disorganized fetal cortex with cytomegalic cells, bizarre nuclei with numerous pseudoinclusions, and a large amount of permanent adult cortex (Figure 1).
DISCUSSION
The phenotype of patients affected with IMAGe association is broad. Intrauterine growth restriction and congenital adrenal hypoplasia are present in all cases (see Table 1). Mild dysmorphic facial features are described, most commonly frontal bossing, epicanthal folds, flattened nasal bridge, small low set ears, smooth philtrum, and micrognathia. Genital abnormalities are also mild, affecting only boys, and usually consist of cryptorchidism or micropenis. Skeletal abnormalities include epiphyseal and metaphyseal dysplasia. Other common findings that have been described in IMAGe association include macrocephaly [8,12,16,17], growth hormone deficiency [10,14], scoliosis [7,8], hypercalciuria [7,8,12,16], and craniosynostosis [7,12].
Literature review of reported cases of IMAGe association
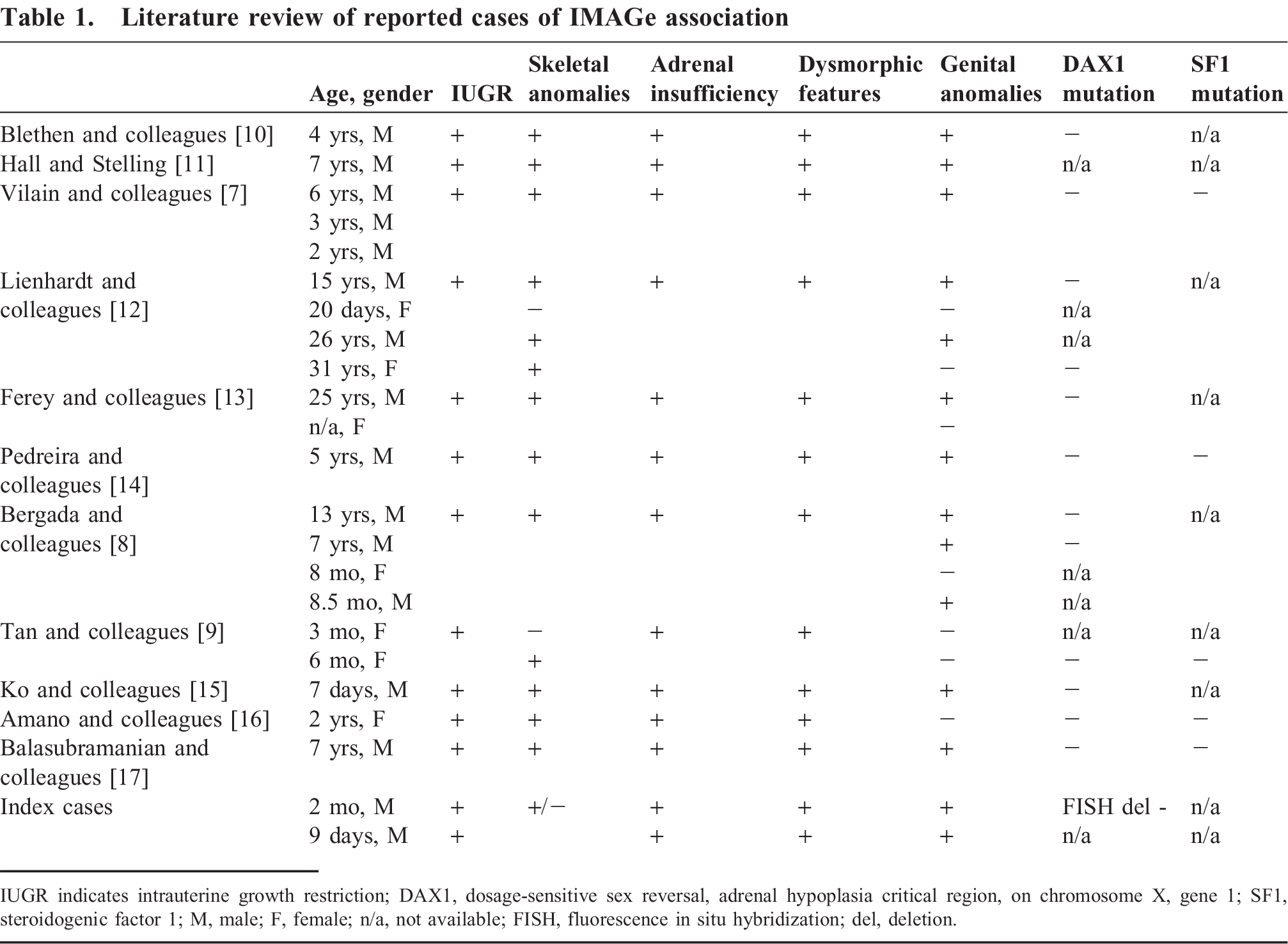
IUGR indicates intrauterine growth restriction; DAX1, dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1; SF1, steroidogenic factor 1; M, male; F, female; n/a, not available; FISH, fluorescence in situ hybridization; del, deletion.
The boy described in Case 1 had many features to support a diagnosis of IMAGe association, including intrauterine growth restriction, mild dysmorphic features (frontal bossing, bilateral epicanthal folds, and flat philtrum) and mild genital abnormalities (left-sided cryptorchidism and penile chordee). He had primary adrenal insufficiency and persistent hypercalciuria. Review of his older brother's autopsy (Case 2), revealed findings which are also suggestive of a diagnosis of IMAGe association, including intrauterine growth restriction, macrocephaly with dysmorphic features, large phallus, and cryptorchidism. Although not formally diagnosed with primary adrenal insufficiency prior to death, autopsy findings of hypoplastic and disorganized adrenal glands suggest the diagnosis. He had transient hyponatremia but was also receiving parenteral nutrition, which can mask underlying electrolyte abnormalities. The boy in Case 2 likely had undiagnosed primary adrenal insufficiency, making him more susceptible to acute adrenal crisis under biological stress, which likely contributed to his demise.
Metaphyseal dysplasia is a feature of IMAGe association which was not found in our case series; however, it is typically a later finding with signs presenting between the ages of 6 months and 7 years [16]. When present, metaphyseal dysplasia is usually found in long bones, although restriction to short, tubular bones has also been described [8,9]. Amano et al. presented the radiological evolution of skeletal abnormalities in IMAGe association and suggested that delayed endochondral ossification may be the only radiological finding present at birth [16]. Metaphyseal dysplasia is often very mild [8,9,16] and may be easily missed, which may have happened in our patient. The boy in Case 1 had only shortened arms and legs, which has been described in numerous reports of IMAGe association [7,12,13,16]. A postnatal skeletal survey, however, showed no abnormalities.
Of interest, both patients in our case series were male, and only 7 of 21 reported cases of IMAGe have been described in females, thus suggesting a slight male preponderance associated with this syndrome. Familial analysis of a large 5-generation Argentinian family with IMAGe association, originally reported by Bergada et al. [8], has demonstrated a maternally imprinted mode of inheritance in IMAGe syndrome, in contrast to the X-linked recessive and autosomal recessive forms of congenital adrenal hypoplasia.
The only description of adrenal gland morphology in IMAGe association to date was described from autopsy findings in a three-month old girl from Tan et al [9]. The adrenal glands were described as hypoplastic and disorganized with cytomegalic cellular morphology in the cortical cells (large cells with eosinophilic, granular cytoplasm, irregular hyperchromatic nuclei, intranuclear inclusions, and cytoplasmic vacuolization) and a well-formed adrenal medulla. These findings are similar to the adrenal gland morphology described for the X-linked recessive, cytomegalic form of congenital adrenal hypoplasia.
The histopathology of the adrenal glands in this case series revealed nodular and hypoplastic adrenal glands with disorganized architecture. Like that previously described, the cortex consisted of large, eosinophilic cells (cytomegaly) with cytoplasmic vacuolization and enlarged nuclei with intranuclear inclusions. Unique to our series is that both cases had foci of permanent adult cortex, suggesting overlapping adrenal histopathologic features between previously described X-linked recessive (cytomegalic cells with lack of permanent adult cortex) and autosomal recessive (diminished permanent adult cortex without cytomegalic cells) congenital adrenal hypoplasia.
In summary, we report two cases of siblings with gross and microscopic features of IMAGe association, a rare disorder characterized by intrauterine growth restriction, metaphyseal dysplasia, congenital adrenal hypoplasia, and genital anomalies. The pathologic findings seen in the adrenal glands suggest IMAGe association falls somewhere in the spectrum between the X-linked recessive and the autosomal recessive forms of congenital adrenal hypoplasia.