
Abstract

INTRODUCTION
Enlarged testes in the pediatric age group may be due to variable causes, ranging from tumors to hyperplasias of the many different testicular components. Some forms of macrorchidism are associated with syndromic entities; others represent reactions to hormonal dysregulations, or specific gene mutations. A common consequence of macrorchidism is precocious puberty. This chapter reviews etiological and phenotypical types of testicular hypertrophy and associated forms of precocious puberty.
Most testicular hypertrophies in childhood are due to testicular tumors (unilateral hypertrophies) or to congenital adrenal hyperplasia (bilateral hypertrophies) [1]. Other examples involve an expansion of testicular parenchyma. To evaluate the amounts of these increases, measurements of testicular parenchymal volume are necessary. Following are some of the available methods which have shown comparable results: Prader's orchidometer, ring orchidometer, ultrasonography, dimensional measurements, or graphic methods [2]. Testicular enlargement may be observed from birth, but it can also begin in infancy. The enlargement can involve 1 or both testes, and is caused by an excessive development of either seminiferous tubules or Leydig cells. The disorder is found in association with chromosomal anomalies, tumors, or endocrine abnormalities. Most cases may be included in 1 of the following presentations: testicular hypertrophy, central precocious puberty, peripheral precocious puberty, and mixed precocious puberty [3].
TESTICULAR HYPERTROPHY
The most frequent presentations of testicular hypertrophy are: congenital Leydig cell hyperplasia, compensatory hyperplasia, benign idiopathic macrorchidism, Martin-Bell syndrome, and testicular hypertrophy associated with juvenile hypothyroidism.
Congenital Leydig cell hyperplasia
Congenital Leydig cell hyperplasia is a relatively frequent condition that may occur in 2 forms: nodular and diffuse [4].
The diffuse form requires quantitative studies for its diagnosis (Figs. 15.1, 15.2). The nodular form is characterized by nodules of Leydig cells in the mediastinum testis or its surroundings (adjacent testicular parenchyma and connective tissue among the ductuli efferentes) (Figs. 15.3, 15.4). These nodules are not encapsulated and show continuity with the normal clusters of Leydig cells in the testicular interstitium.

The differential diagnosis of nodular Leydig cell hyperplasia includes intratesticular adrenocortical remnants and bilateral Leydig cell tumors. Adrenal remnants are encapsulated (except those arising in the adrenogenital syndrome) and consist of radial cell cords. These cells have vesicular nuclei with small nucleoli. Leydig cells tumors usually are unilateral, located more deeply in the testicular parenchyma, and appear poorly delimited. The latter feature also occurs in nodular Leydig cell hyperplasia, adding difficulties to the differential diagnosis. Leydig cell tumors are rare in newborns and, when they appear during infancy, produce large amounts of androgens. This induces precocious maturation of the adjacent seminiferous tubules, giving rise to a syndrome of macrogenitosomia.
Congenital Leydig cell hyperplasia is caused by increased hCG which, in some circumstances, enters the fetal circulation [5]. A large number of pregnant diabetic women, mainly those with hypertension, develop edematous placental villi and increased vascular permeability with subsequent entrance of hCG into the fetal compartment. Congenital Leydig cell hyperplasia disappears rapidly in the first months of extrauterine life.
Congenital Leydig cell hyperplasia has been reported in triploid fetuses [6], infants with Beckwith-Wiedemann syndrome [7], leprechaunism, nonimmune hydrops fetalis [8], Rh isoimmunization [4], and several complications of pregnancy [9].
Beckwith-Wiedemann syndrome (BWS) is the best known and most frequent overgrowth syndrome, with an estimated prevalence of 1 in 12 000 live births [10]. Originally described by Beckwith in 1963 [11] and later by Wiedemann [12] in 1964, this syndrome is characterized by macroglossia, prenatal or postnatal overgrowth, omphalocele and other abdominal defects (early signs). This is followed by neonatal hypoglycemia, which demands early recognition and treatment to avoid severe neurological damage. Additional features are visceromegaly and variable hemihypertrophy or limb hypertrophy [10,13]. Multiple organs, including the adrenal cortex, kidneys, pancreas, and gonads, show overgrowth features. This is a cancer predisposition syndrome, leading to tumors in infants and children (≈7.5% of patients) but also in adults. Multiple genes appear to play a role in BWS. The impacts of genomic imprinting and assisted reproductive techniques in its pathogenesis have been emphasized for several years [14,15]. Dysregulation of imprinted growth regulatory genes at the 11p15.5 region has been documented. Most adult BWS patients show anomalies in testicular function or decreased spermatogenesis. These have been related to cryptorchidism or endocrine alterations, but a primary testicular anomaly cannot be ruled out [16].
Compensatory hypertrophy of the testis
Compensatory testicular hypertrophy was first reported by Laron and Zilka [17] in 1969 in unilateral cryptorchid patients. It has also been observed in monorchidism [18], varicocele [19], and in some testes after injury. This hypertrophy is characterized by increased volume of the scrotal testis, defined as more than 2 standard deviations above the corresponding size for age in normal children.
Compensatory hypertrophy persists during infancy and puberty, and ceases when puberty is completed. The size of the formerly hypertrophic (compensating) testis in adulthood is normal or slightly increased [20,21].
The degree of compensatory hypertrophy is determined by 3 factors: (a) the amount of testicular volume reduction; (b) the age at the time of the injury; and (c) the histology of the contralateral descended testis [18].
Compensatory hypertrophy seems to be caused by abnormal pituitary control followed by increased follicle-stimulating hormone (FSH) secretion, and it is taken for granted that the hypertrophied testis is otherwise normal.
In monorchidism, it is assumed that the absent testis was normal during fetal development [22] and later underwent progressive atrophy. Only when testicular mass reduction reaches 50%, which occurs shortly before birth, the endocrine feedback mechanism becomes abnormal, increasing FSH secretion leading to accelerated growth of the contralateral testis (Figs. 15.5, 15.6).
In cryptorchidism, 2 circumstances should be taken into account: testicular mass reduction is not as pronounced as in monorchidism, and the contralateral scrotal testis may also be abnormal. Therefore, hypertrophy of the contralateral testis in cryptorchidism is usually less prominent than in monorchidism.
Age is a decisive factor for the development of hypertrophy. Most compensatory hypertrophies develop from birth to 3 years of age. At this time, the testicular volume may be twice the normal volume when the contralateral testis is absent [23].
In infancy, monorchid infants with compensatory testicular hypertrophy have low levels of inhibin B, and high levels of FSH. This suggests that compensatory testicular hypertrophy does not prevent testicular insufficiency in adulthood. An early endocrinological evaluation, combined with early sperm analysis, is advised for males with compensatory hypertrophy at puberty [24].
Idiopathic benign enlargement
Some prepubertal and pubertal patients show pronounced, bilateral asymptomatic [25–27], or unilateral [28] testicular hypertrophy. This disorder is considered the morphological expression of a peculiar sensitivity of testicular parenchyma to hormonal stimulation. In most patients the clinical presentation coincides with the onset of puberty, or immediately before (Figs. 15.7, 15.8).
Before diagnosing this entity, several disorders should be excluded: bilateral testicular neoplasia (germ cell tumors, stromal tumors, leukemia, or lymphoma); adrenal rest tumors; X-linked mental retardation; hypothyroidism; and, idiopathic or cerebral precocious puberty.
In morphometric studies, testicular enlargement is most often the result of excessive lengthening of the seminiferous tubules. The main histological findings are Sertoli cell hyperplasia and deficient spermatogenesis. Elevated FSH, detected in some cases, might be the cause of excessive Sertoli cell proliferation. This increases the diameter and length of the seminiferous tubules, giving rise to macrorchidism [29–31]. In some cases, the underlying mechanism is unclear, with normal sequencing analysis of ten exons of the FSHR [32]. Unilateral presentations may represent a well known phenomenon: asynchronous pubertal development of testes. No increased risk for malignancy is reported in association with benign testicular enlargement.
A peculiar situation of testicular enlargement was observed in a 14-year-old asymptomatic male whose testicular parenchyma on sonogram had conspicuous diffuse lobulations, reminiscent of fetal renal lobulations seen in newborns [33].
Fragile X-chromosome syndrome: Martin-Bell syndrome
The fragile X-chromosome syndrome is the best known form of inherited intellectual disability, with an incidence of 1 in 4000 males and 1 in 6000 females [34]. The syndrome was first reported in 1943 by Martin and Bell [35] in multiple male members of a family. In 1969, Lubs [36] found a break on the X chromosome in these patients. In 1970, Hecht and Lovrien [37] used “fragile site” to name this abnormality, and Martin-Bell syndrome was thereafter known as fragile X syndrome (FXS). Multiple presentations of this syndrome were described in the ensuing 2 decades [38–46]. In 1991, the responsible gene was mapped to locus q27.3 of chromosome X, which is now referred to as the fragile X mental retardation 1 (FMR1) gene [47].
Causes of peripheral precocious puberty

The cause in almost all cases of FXS is a trinucleotide CGG repeat in the 5′ untranslated region of the FMR1. In humans the number of repeats is highly polymorphic. Normally there are between 6 and 54 repeats, with 29 or 30 repeats being the most common allele [48–50]. When the number of repeats increases to between 60 and 200, it is called a premutation allele. In males this premutation is asymptomatic, although in females it is associated with a high incidence of dizygous twin pregnancies (4 to 5 times that of normal women), and a higher frequency (20%) of premature ovarian failure [48–50]. Patients with this permutation are at high risk for developing fragile X-associated tremor ataxia syndrome (FXTAS). This neurological condition affects males and females over 50 years of age, with action tremor and ataxia, neuropathy, cognitive decline, autonomic dysfunction, and parkinsonism [51]. Premutation alleles of the FMR1 gene contribute to the FXS phenotype through genetic instability and expand into the full mutation during germline transmission [52]. These carriers have a high risk of developing fragile X-associated tremor/ataxia syndrome.
When the number of repeats exceeds 200, it is considered a full mutation. This leads to hypermethylation and chromatin condensation, preventing the binding of specific transcription factors and altering the basal transcriptional machinery. The result is transcriptional gene silencing. As a consequence, the gene product, fragile X mental retardation protein (FMRP) is absent, causing FXS [53]. FMRP is a cytoplasmic 70- to 80-kDa RNA-binding protein that regulates translation of a number of mRNAs whose protein products are important for synaptic development, maintenance, and plasticity. In its absence many synaptic proteins are dysregulated [54]. The inheritance is dominant, X-chromosome-linked, with low penetrance in females and variable expression in males [55].
Clinically, detection of the fragile X phenotype in the prepubertal period is difficult. A recent pilot study in newborns identified an unexpected high incidence (1 in 730) [56]. Patients show mild to moderate mental retardation and autism with hyperkinetic behavior [57]. Physical characteristics include macrocephaly (prominent forehead and ears, long face, prognathism), macrorchidism, and multiple connective tissue abnormalities (hypotonia, flat feet, mitral valve prolapse, soft skin, hyperextensible finger joints, and high arched palate). Males with full fragile X mutation also have intellectual disability with low IQ and symptoms in common with autism spectrum disorder such as delayed speech and language, impaired theory of mind, social and emotional processing, irritability, hyperactivity, and attention deficits [58]. The prevalence and severity of FXS in females is less severe because of lyonization of the X chromosome.
Remarkable findings occur in tissues with higher expression of FMRP, such as neurons in the hippocampus and cerebellum, and testes [59]. Neuroimaging reveals increased size of the cerebellum, amygdala and hippocampus, larger caudate nucleus, hypoplasia of vermis, and ventricular abnormalities [60]. The right hemisphere is more affected than the left. Autopsy studies also demonstrate abnormalities in hippocampus, cerebellar vermis, and in the frontal cortex. Focal thickening of CA1 in the hippocampus, irregularities in the dentate gyrus, and intranuclear eosinophilic inclusions in neurons and astrocytes of these areas have been observed. Inclusions measure 2 to 5 microns and are positive for ubiquitin; they fit the description for intranuclear inclusions in FXTAS [61]. Similar inclusions have been observed in the frontal cortex. These inclusions are only present in FMRP-immunoreactive cells and are observed in patients with both premutation and full mutation FMR1 [62]. Located in the cerebellar vermis are decreased, misplaced, and abnormally oriented Purkinje cells. These histological findings overlap with those reported in cases of autism and schizophrenia [63]. The lack of FMRP leads to dendritic spine dysmorphogenesis and impaired synaptic plasticity.
Macrorchidism (testicular volume higher than 25 ml in adults) is present in more than 75% of affected adult patients. The average testicular volume in these patients is 70 ml, a testicular size 4 times larger than that of the normal testis. In addition to the characteristic mental retardation and facial dysmorphism (large spread ears that may require surgical correction), a large penis is noticeable from the first years of life through adulthood.
Testicular enlargement probably begins during fetal life [64]. In infancy, testes are enlarged, scrotal pouches are more developed than normal, and genital pigmentation appears early [65]. This exaggerated genital development is difficult to explain, since the function of the hypothalamic-pituitary-gonadal axis is normal. Some patients show slightly increased FSH levels, suggesting an abnormally high sensitivity to this hormone [65].
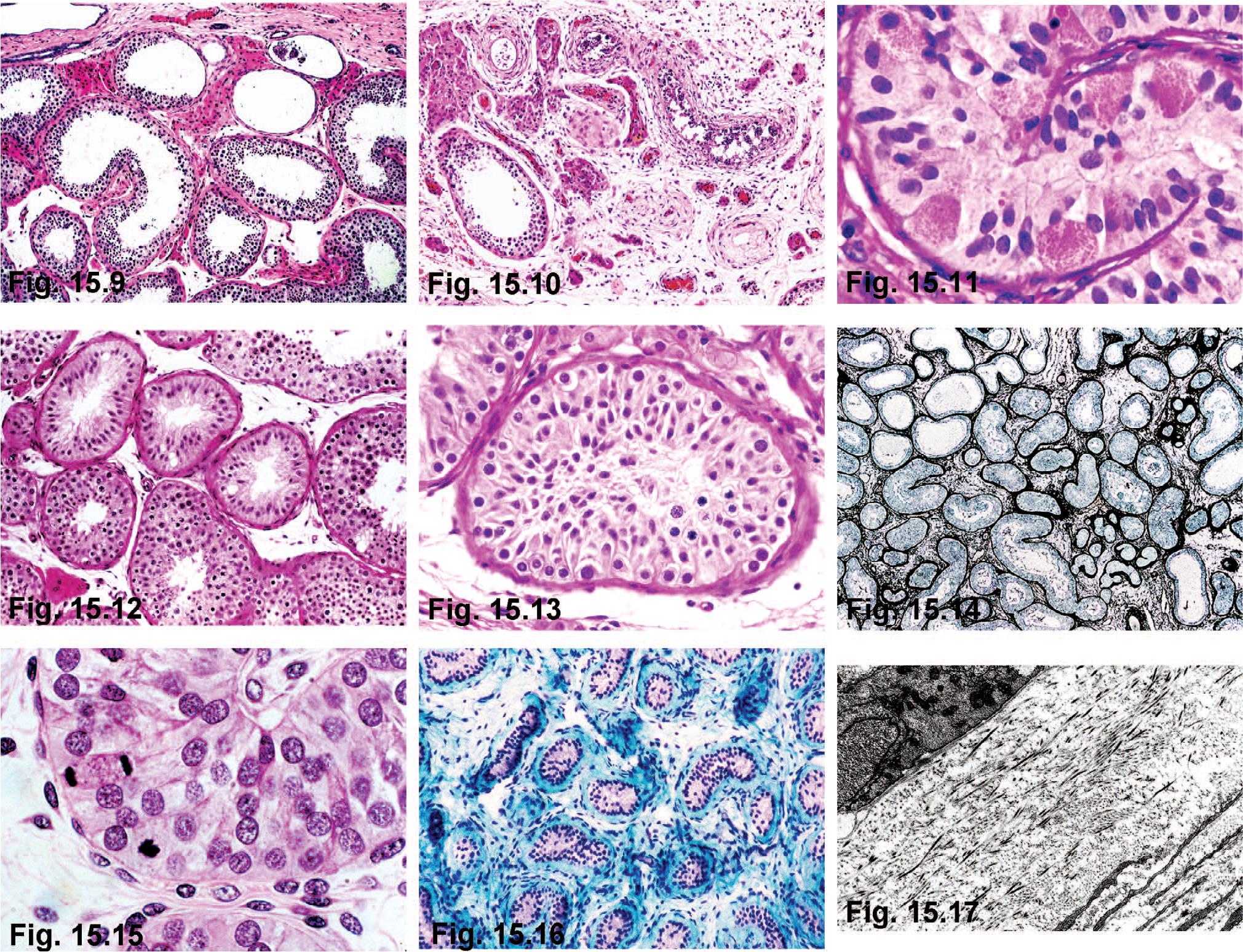
Testicular biopsies in some adult patients have been normal or have nonspecific changes, such as interstitial edema and reduced spermatogenesis [66]. In other cases, severe lesions include marked tubular ectasia (Fig. 15.9), atrophy of the seminiferous epithelium (Fig. 15.10), granular changes in Sertoli cells (Fig. 15.11), mixed atrophy with or without Sertoli cell hyperplasia (Figs. 15.12, 15.13), and Sertoli cell nodules (Fig. 15.14). The increase in testicular size may be due to excessive Sertoli cell proliferation subsequent to the lengthening and coiling of the seminiferous tubules [30]. The consistently low number of spermatozoa in spermiograms might be explained by abnormal Sertoli cell maturation with deficient spermatogenesis. Meiotic anomalies have been excluded [67]. Intranuclear inclusions like those seen the nervous systems in both FXS and FXTAS have been observed only in testes of patients with FXTAS. In contrast with FXS patients, those with FXTAS usually have atrophic testes. Ubiquitin-positive inclusions are found only in myoid and Leydig cells [68].
The following diagnostic approaches have been proposed for this frequent anomaly: (a) cytogenetic analysis of patients with mental retardation and no or only slight dysmorphic signs; (b) Southern blot analysis in patients with family history or several signs of FXS; (c) PCR studies if screening of a greater population is required; and (d) immunohistochemistry with monoclonal antibodies to detect FMR1 protein [69–71]. Southern blotting is the gold standard for FMR1 molecular diagnostic methods [72,73].
In fragile X patients, genotype correlates with phenotype. Abundant CGC unit repeats correlate with the most severe forms of phenotype. On the other hand, nonmethylated CGC repeats show little or no repercussion [74].
Two therapeutic approaches are currently under investigation for patients with FXS: reactivation of the FMR1 gene, and compensating for the lack of FMRP [75].
The terms “negative fragile X Martin-Bell syndrome” or “X-linked mental retardation macrorchidism” (MRMO or XLMR+MO) describe patients with the characteristic phenotype of Martin-Bell syndrome but without fragile X. The gene responsible for this different syndrome has been mapped to the Xq12–q21 region [76].
Isolated patients with fragile X-chromosome have developed testicular torsion [77] and neoplasms (testicular or colonic) [78].
Other testicular hypertrophies
Testicular hypertrophy has been associated with glandular disorders including FSH-secreting pituitary adenoma [79], hyper- and hypoprolactinemia, and hypothyroidism [80,81]. The most frequent association is with hypothyroidism.
Testicular hypertrophy associated with hypothyroidism
Children with untreated hypothyroidism usually have a delayed onset of puberty. The association of juvenile hypothyroidism and isosexual precocious puberty is the van Wyk-Grumbrach syndrome (VWGS), initially described in 1960 [82]. It is more frequent in females than in males. Children with hypothyroidism frequently have testicular enlargement without virilization and delayed bone age [81,83,84]. Approximately 80% have macrorchidism [85,86]. The degree of testicular change is directly proportional to the duration of hypothyroidism. Most patients show elevated FSH and prepubertal luteinizing hormone (LH) serum levels [87,88]. Serum testosterone levels are within normal limits; the response to FSH and LH stimulation is abnormal, and no pulsatile LH is released [89].
Testicular biopsies before puberty have accelerated development, with pubertal maturation of the seminiferous tubules, increased numbers of Sertoli cells, minimal changes in spermatogenesis, and absence of Leydig cell development.
Testicular biopsies in adults with untreated juvenile hypothyroidism have hyalinized tubules, peritubular and interstitial fibrosis, and scant Leydig cells [90,91] (Figs. 15.15–15.17). Pubertal and adult patients with chronic untreated hypothyroidism usually have abnormal testicular biopsies and spermiograms [92].
Macrorchidism is reduced as soon as hormonal replacement is administered [87,93–95].
Different hypotheses have been proposed to explain the pathogenesis of this testicular hypertrophy, for example: (a) the low level of T3 and T4 induces an increase of thyrotropin-releasing hormone (TRH) secretion, resulting in increased levels of TSH, gonadotrophins and prolactin. Elevated FSH would induce testicular hypertrophy [96,97]; (b) direct TSH action on the testis, mediated by FSH receptors in testicular cells, is due to the similar glycoprotein structure of the hormone receptors [98,99]; (c) deficit in thyroid hormones, normally required for testicular maturation, result in the maintenance of Sertoli cell proliferation, giving rise to increased testicular volume [100–103]; (d) loss-of-function mutations in the immunoglobulin superfamily member-1 (IGSF1) gene cause an X-linked syndrome of central hypothyroidism [104]; and (e) other hypotheses including hyperprolactinemia and abnormalities in the steroid metabolism of testicular cells.
Testicular hypertrophy secondary to FSH-secreting pituitary adenoma
Most pituitary adenomas (80%–90%), even though they are considered nonfunctioning, secrete variable amounts of FSH. But only exceptionally do they have clinical manifestations [105,106]. Functioning pituitary adenomas with FSH secretion during infancy and puberty, give rise to variable clinical symptoms. Once the most frequent causes of macrorchidism have been ruled out, this diagnosis is suggested by a discrepancy between normal or elevated serum levels of FSH and decreased LH, as well as by the detection of elevated inhibin B levels. In males, FSH acts exclusively on membrane-bound FSH-receptors on Sertoli cells, stimulating spermatogenesis through increased production of paracrine factors and androgen-binding protein. Histological studies suggest that testicular enlargement is due to increased length of the seminiferous tubules [79]. After surgical removal of the adenoma, serum FSH normalizes and testicular volume decreases [107].
PRECOCIOUS PUBERTY
Puberty is a period of changes that lead to sexual maturation and active reproductive function. Puberty is preceded by adrenal gland maturation known as adrenarche, which takes place at 7 to 8 years of age. Pubertal development is a sequential process and starts in males at an average of 11.2 years of age. Numerous neuroendocrine factors control the beginning of puberty, including adrenergic and dopaminergic neurotransmitters, endogenous opioids, and melatonin secreted by the pineal gland [108]. These factors stimulate the hypothalamus to produce a pulsatile release of gonadotropin-releasing hormone (GnRH), increasing pituitary gonadotropin secretion, and stimulation of gonadal function.
Precocious puberty is defined by the appearance of secondary sex characteristics at an age 2.5 standard deviations below the average of a given population. From a practical viewpoint, precocious puberty may be diagnosed when it occurs before 8 years of age in females and nine years of age in males [109]. The incidence of this disorder is between 1 in 5000 and 1 in 10 000, with a female-to-male ratio of more than 20:1. In males, the first sign of pubertal development is rapid testicular enlargement, followed by development of axillary and pubic hair, increase in penile size, and acceleration of body growth [110].
Male precocious puberty may be classified in 3 major groups depending on the hypothalamic-pituitary-gonadal axis function, as follows: (1) central precocious puberty or gonadotropin-dependent precocious puberty (GDPP), also called “true precocious puberty,” which results from activation of the hypothalamic-pituitary-gonadal axis; (2) peripheral, or gonadotropin-independent precocious puberty (GIPP), mediated by release of sex steroid hormones from adrenal glands, testis, and extratesticular germ cell tumors; or (3) mixed precocious puberty begins as peripheral precocious puberty and transforms into gonadotropin-dependent precocious puberty after inducing hypothalamic activation.
Central precocious puberty
Central precocious puberty (CPP) recapitulates the physiological pubertal development but at a premature chronological age. The first manifestation is an increase in the volume (>4 ml) or length (>2.5 cm) of testes. The disorder is isosexual. The diagnosis is easily made by detection of elevated serum gonadotropin levels (both basal and in response to GnRH test), associated with increased serum testosterone levels, elevated LH-to-FSH ratio, or increased LH and FSH levels after stimulation with GnRH agonists. In some cases, nocturnal measurement of LH secretion, to determine LH pulses before performing a dynamic test, shows a pubertal pattern. Clinically, the high concentration of steroids determines an accelerated growth and bone maturation. The rapid growth ends with the premature fusion of epiphyses, resulting in an adult with short stature.
This is the most common form of precocious puberty, accounting for more than 50% of cases in males, and most of female cases. The age of presentation is from 4 to 8 years of age [111].
The cause of CPP is known in 60% of cases: 40% have nervous system (NS) lesions; the remaining cases are represented by genetic and idiopathic variants [112]. In males, introduction of computed tomography (CT) scans and magnetic resonance imaging techniques has greatly enhanced identifying the causes [113–115].
CPP secondary to nervous system lesions
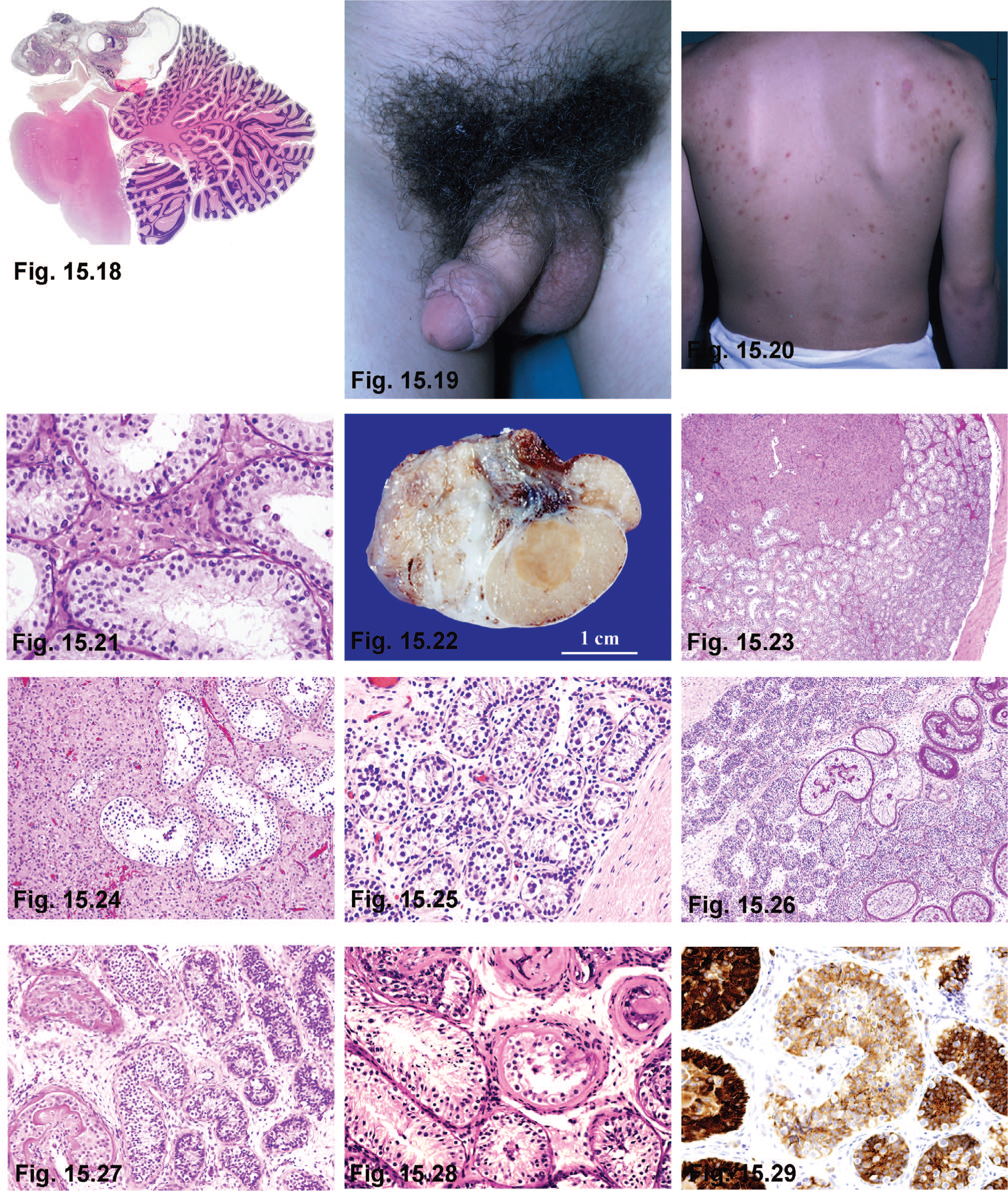
The nature of NS lesions differs from 1 case to another, but in all, the specific areas involved include posterior hypothalamus (medial eminence and tuber cinereum), mammillary bodies, floor of the third ventricle, or the pineal gland [116,117]. The most frequent causes include several tumors, including hypothalamic (astrocytomas, ganglioneuromas, gangliogliomas, craniopharyngiomas, cysts of the third ventricle, and suprasellar arachnoid cysts) [118–120]; pineal cysts [121]; hamartomas (gangliocytomas) of the tuber cinereum and the mammillary body; tumors of the pineal gland (teratomas and pinealomas) (Figs. 15.18–15.20); tumors of the optic nerve (gliomas), cerebral and cerebellar astrocytomas; and granular cell tumor of the neurohypophysis [122]. Other causes include post-partum and accidental cerebral traumas which stimulate extrahypothalamic areas responsible for hypothalamic activation [123–125]; infections, including meningitis, encephalitis, tuberculosis, toxoplasmosis and syphilis; cerebral malformations including hydrocephalia, microcephalia, craniostenosis [126], pituitary duplication, and midline defects [127]; and inherited phakomatoses such as neurofibromatosis and tuberous sclerosis. Children with neurofibromatosis type 1 often develop optic pathway tumors [128].
Additional contributing factors are selective hypothalamic-hypophyseal irradiation [129], prophylactic cerebral irradiation in children with acute lymphoblastic leukemia [130], and irradiation of cerebral tumors outside of the hypothalamic-hypophyseal region [131,132].
Treatment of peripheral precocious puberty must be considered. An important observation derived from the use of CT scan and magnetic resonance is the presence of a high number of hamartomas in children with precocious puberty [133–135]. These lesions represent gangliocytomas, and they appear as multiple small foci located in the hypothalamus, between the anterior portion of the mammillary body and the posterior part of the tuber cinereum. Immunohistochemical, ultrastructural and genetic studies have shed light on how these lesions lead to precocious puberty. Immunohistochemical staining of GnRH-positive neurons in these hamartomas supports the notion that these neurons generate GnRH pulses; GnRH enters the hypophyseal portal system and reaches pituitary gonadotropic cells [136]. Other hamartomas associated with CPP show immunohistochemical reactivity to transforming growth factor alpha (TGFα) protein (and express its mRNA), and to TGFα and epidermal growth factor (EGF) receptors [137], which could also be involved in development of precocious puberty. Recently, special importance has been given to kisspeptin (Kiss1). An activating mutation (P745) in the Kiss1 gene, encoding G protein-coupled receptor 54 (GPR54) ligand (Kisspeptin) has been reported in some instances of precocious puberty [138,139].
Precocious puberty due to brain tumors appears in advanced stages of tumor development, and its clinical symptoms are preceded by other cerebral symptoms, including hydrocephaly, papillary edema, and psychic abnormalities [140]. The same occurs when precocious puberty is secondary to cerebral inflammation or malformations.
Pineal tumors are rare in children, but 30% of them cause precocious puberty, mainly in males. The most conspicuous are teratomas and tumors extrinsic to the pineal gland that destroy its parenchyma. Suppression of the antigonadotropic action of this gland causes precocious puberty [141]. Tumors consisting of pinealocytes secrete great amounts of melatonin, which delays the onset of puberty.
Idiopathic central precocious puberty
The cause of CPP cannot be ascertained in approximately 80% of female and 40% of male children. The difference between genders might be attributed to the higher sensitivity of female gonadotropic cells to GnRH stimulation. Etiologies include genetic and ethnic factors, pediatric obesity, family history of psychosocial stress, and adoption [142]. In addition, degradation products of chemicals such as dichlorodiphenyltrichloroethane (DDT) have been implicated [143]. Idiopathic precocious puberty is familial in nearly half of the cases, with puberty starting after 7 years of age in most males. Inheritance may be autosomal recessive or sex-linked with variable penetrance [144]. Recently, genetic causes have been considered important in some cases of idiopathic CPP [145–148]. Other pathologies associated with idiopathic CPP include: intrauterine growth retardation [149], Silver-Russell syndrome, bilateral retinal degeneration, epilepsy, cryptorchidism, and inguinal hernia [150]. Gonadotropin-releasing hormone agonists are the treatment of choice [151].
Peripheral precocious puberty
Peripheral precocious puberty (PPP), also known as precocious pseudopuberty, is far less common than CPP. PPP may be due to primary testicular causes, different endocrine extratesticular abnormalities, or result from hormonal treatments. Among the primary causes, the following disorders have been reported: familial testotoxicosis, functioning testicular tumors, excessive aromatase activity, and Leydig cell hyperplasia with focal spermatogenesis. Secondary causes involve other endocrine glands, and include several adrenal cortical abnormalities (congenital adrenal hyperplasia, virilizing carcinoma of the adrenal gland, and Nelson syndrome), and lesions secondary to human chorionic gonadotropin (hCG)-secreting tumors. Hepatoblastomas are responsible for 50% of such cases. Testicular, retroperitoneal, mediastinal, and pineal germ cell tumors contribute the remainder [109,152–154]. The best-known iatrogenic cause of precocious puberty is long-term administration of hCG [109].
Familial testotoxicosis (GIPP) or familial male-limited precocious puberty
Familial male-limited precocious puberty (FMPP) is a form of male precocious puberty characterized by early maturation of Leydig cells and spermatogenesis, in absence of pituitary gonadotropic stimulation. In 1993, Shenker and colleagues [155] reported the first constitutively activating mutation of the gene encoding the LH chorionic gonadotropin (LH/CGR) receptor. Children, usually from 1 to 4 years of age, present signs of puberty, including rapid virilization, accelerated growth with eventual closure of epiphyses, and short stature in adulthood. Although their testes are enlarged, testicular size does not correlate with degree of virilization (Fig. 15.21).
This disorder is considered an autosomal dominant, primary testicular abnormality, limited to males [156–158], due a constitutive activating mutation of the LH/CGR gene [159–162]. Recently, a novel C617Y mutation of this gene has been reported [163]. The LHR gene consists of 11 exons and has been mapped to 2p21 [164]. Hormone assays show elevated serum testosterone, low dehydroepiandrostenedione (DHEA), androstenedione, 17-hydroxyprogesterone, GnRH, and LH; absence of pubertal LH pulsatile pattern [155], and elevated inhibin B before the normal age of puberty [165]. The gonadotropin response to GnRH stimulation is negative. Ultrastructural studies reveal adult development of Leydig cells and complete spermatogenesis, although with many spermatid morphological anomalies [166]. In some patients, LHR mutation induces nodular Leydig cell hyperplasia [167,168].
Patients do not respond to treatment with GnRH analogs, used to treat central precocious puberty. Therapy with ketoconazole is effective, but hepatotoxicity may develop and some patients also suffer an “escape” phenomenon (secondary central precocious puberty). This requires additional therapy with GnRH agonists [169]. A proposed alternative to GnRH analogs is the administration of cyproterone acetate or aromatase anastrozole inhibitor [170,171] and bicalutamide, a nonsteroidal antiandrogen [172]. Inactivating mutations of GnRH cause male undermasculinization due to absence or hypoplasia of Leydig cells [173].
Precocious pseudopuberty secondary to functioning tumors
The following tumors may produce precocious pseudopuberty: Leydig cell tumor, sex cord tumor, virilizing carcinoma of adrenal cortex, and extratesticular hCG-secreting tumor.
Leydig cell tumor—These tumors may give rise to precocious puberty in children (Fig. 15.22). Testicular enlargement is caused in part by the tumoral mass, but is mainly due to the maturation of the adjacent seminiferous tubules induced by androgen secretion. The presentation is unilateral in most cases, and the contralateral testis is only rarely enlarged [174]. An activating mutation of the LH receptor (LHR) gene Asp578 His has been detected in several patients with Leydig cell tumors [175]. The onset of precocious pseudopuberty is variable, but in most cases it occurs in the first 5 years of life [176]. In some cases, probably because of an early diagnosis, tumors cause precocious tubular maturation but no precocious pseudopuberty [177]. Ultrasonography scanning, selective venous sampling, and the hCG stimulation test help to diagnose the tumor [178]. In children, the same clinical manifestations described for functioning Leydig cell tumor have been seen in Leydig cell hyperplasia. Patients show precocious pseudopuberty and ipsilateral testicular enlargement [179]. On histologic examination, hypertrophic Leydig cell nodules are located among or surrounding seminiferous tubules with complete spermatogenesis. These Leydig cells lack Reinke's crystals and do not compress seminiferous tubules. There is a clear delimitation between seminiferous tubules with spermatogenesis and those showing an infantile maturation pattern (Figs. 15.23–15.25). The differential diagnosis between Leydig cell hyperplasia and Leydig cell tumors causing precocious pseudopuberty is not easy. Open testicular biopsy has been recommended, and if a Leydig cell tumor is diagnosed, tumor enucleation is advised [180,181]. Whether Leydig cell hyperplasia with focal spermatogenesis in children is an early stage of Leydig cell tumor or a different entity remains controversial.
Sex cord tumors—Large cell hyalinizing Sertoli cell neoplasia (Figs. 15.26–15.29), and large Sertoli cell tumor with calcifications may induce bilateral testicular enlargement. This is caused by tumor growth and by the precocious tubular maturation. Additional signs of precocious pseudopubertyare a picture of isosexual muscle development, axillary and pubic hair and heterosexual gynecomastia. The tumor cells may stimulate Leydig cells to synthesize androgens. Tumor cells themselves then aromatize these androgens into estrogens, and both androgens and estrogens are responsible for clinical symptoms [182,183] (e.g., see aromatase excess syndrome). These tumors are frequently observed in patients with Peutz-Jeghers syndrome [184,185] and those with Carney complex [186].
Adrenocortical tumors—In childhood, most patients with virilizing tumors of the adrenal cortex have small testes, but exceptional examples with testicular hypertrophy have been observed [187]. The seminiferous tubules may have developed under adrenal androgen effect [188]. In untreated or inadequately treated congenital adrenal hyperplasia, both of the testes may enlarge because of “tumoral” growth of cells similar to those of the adrenal cortex [189]. A similar condition may be observed in Nelson syndrome patients.
Extratesticular hCG-secreting tumors—A mildly increased testicular size has been observed in patients with paraneoplastic precocious pseudopuberty secondary to hepatoblastoma [190–192]. Other extratesticular (retroperitoneum, mediastinal, basal ganglia, pineal or suprasellar region) hCG-secreting germ cell tumors may also cause testicular enlargement [193–198]. However, some patients show precocious pubertal maturation that can be either nodular or diffuse. The nodules consist of hyperplastic Leydig cells and seminiferous tubules that may have complete spermatogenesis. Outside the nodule, seminiferous tubules maintain their prepubertal pattern. In some cases, only diffuse Leydig cell hyperplasia is observed [199].
Precocious pseudopuberty secondary to excess of aromatase activity
Biosynthesis of C18 estrogens from C19 androgens takes place in 3 consecutive oxidative reactions, and is catalyzed by an enzymatic complex called estrogen synthetase or aromatase [200]. This complex consists of 2 components, P450arom (a product of the CYP19 gene, located at 15p21.1) [201], which binds a C19 substrate and catalyzes oxygen insertion, leading to formation of C18 estrogens; and NADPH-cytochrome P450 reductase. The latter is a ubiquitous flavoprotein that adds reducing equivalents to any form of cytochrome P450 it binds.
Aromatase is located in the smooth endoplasmic reticulum of estrogen-producing cells, and is expressed in placenta, granulosa cells, Sertoli cells, Leydig cells, adipose tissue, and several regions of the nervous system including, hypothalamus, amygdala and hippocampus. In a patient with Silver-Russell syndrome, phenotype testicular enlargement developed secondary to increased aromatization. Contrary to what is usually seen in this syndrome, the picture of precocious puberty was not central but due to Sertoli cell hyperplasia [202].
Aromatase excess syndrome—Aromatase excess syndrome is an autosomal dominant disorder characterized by excessive conversion of androgens into estrogens. Males with this condition develop heterosexual precocious pseudopuberty with gynecomastia, whereas females develop isosexual precocious pseudopuberty with macro-mastia. In both genders, children show advanced bone age with short stature in adulthood due to the estrogen-induced premature closure of epiphyses. Most males with aromatase excess are fertile and have normal libido [203]. The increased androgen aromatization leads to high serum estradiol and estrone levels, with low testosterone and androstenedione and suppressed secretion of gonadotropins. Usually, the inhibitory effect of estrogens on testicular function is milder than in patients with estrogen-producing tumors or in patients who received treatment with exogenous estrogens [204]. Treatment with aromatase inhibitors is effective [205].
Aromatase deficit—This is a rare autosomal recessive disorder first reported in humans in 1991 [206]. It results from several mutations in the coding region of the CYP19A1 gene, leading to a decrease or loss of enzymatic function and subsequent estrogenic deficit. Most patients are homozygous for inactivating mutations because they are sons of consanguineous parents. Other patients are compound heterozygous born from nonrelated parents.
In both genders, the first symptom appears in the pregnant mother who becomes progressively virilized due to placental inability to aromatize androgens. In female fetuses, excessive exposure to androgens in utero leads to ambiguous external genitalia. In puberty, a normal adrenarche is present; however there is primary amenorrhea, absence of mammary development and progressive hyper-gonadotropic hypogonadism with hyperandrogenism.
In males, aromatase deficiency in childhood is not very significant. As a matter of fact, children with aromatase deficit show normal FSH levels, and their gonadotropin response to GnRH stimulation is also normal [207]. Most cases are diagnosed during puberty [208]. The most important symptom is a continuous linear growth in adulthood. Estrogens have an important effect on growth plates. In the absence of estrogens, growth plate fusion does not occur, despite high levels of androgens, and patients develop a eunuchoid habitus [209]. Other anomalies include genu valgum, osteopenia, osteoporosis and delayed osseous age. Many male patients are obese, with dyslipidemia, hyperinsulinemia, acanthosis nigricans, and diabetes mellitus. Because estrogens play an important role in pituitary regulation in adult males, aromatase deficit causes low libido and infertility [210]. Hormone analyses reveal undetectable estradiol and estrone levels, and elevated gonadotropins, suggesting lack of negative feedback of both FSH and LH by estrogens [211]. Serum levels of androstenedione and testosterone may be normal or increased [212].
Testes have been small in some cases. Cryptorchidism [213] and macrorchidism with normal testicular consistency [214] have been observed in other cases. Many patients have severe oligozoospermia with 100% of immotile spermatozoa.
A phenotype similar to that of men with aromatase deficit is estrogen resistance caused by disruptive mutations of the estrogen-receptor gene. These patients show normal serum testosterone levels and increased levels of FSH, LH estradiol, and estrone [215]. Spermiogram reveals low numbers of spermatozoa (25 million/ml) with decreased viability (18%) [216].
Estrogen therapy is effective and achieves epiphyseal closure, but this treatment does not improve spermatogenesis [213,214,217]. The role of estrogens in male infertility is still under discussion [218,219].
DAX-1/NROB1 mutations with precocious puberty Mutations in the dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1 (DAX1), also called nuclear receptor subfamily 0, group B, member 1 gene (NROB1), usually produce delayed or incomplete puberty associated with partial gonadotropin deficiency. Rare cases develop gonadotropin-independent precocious puberty [220]. The mechanism of precocious puberty is probably multifactorial, and several hypotheses have been proposed: NROB1 is involved in pubertal control of the gonadal axis; the stimulus exerted by ACTH on melanocortin receptors in Leydig cells; and the overexpression of testicular steroidogenesis activators by the loss of transcriptional repression [221,222].
Mixed, peripheral, and central precocious puberty
The most characteristic example of mixed precocious puberty is the McCune-Albright syndrome (MAS). This was first described separately by McCune in 1936 [223] and by Albright and colleagues [224] in 1937. This is a rare disorder, characterized by polyostotic fibrous dysplasia, “cafe au lait” skin pigmentation, and autonomous endocrine hyperfunction. Its incidence is estimated from 1 in 100 000 to 1 in 1 000 000. Sexual precocity is frequent in females, but is present in only 15% of males. Male patients show testicular enlargement, prepubertal size of the penis, and absence of axillary and pubic hair. Although testicular enlargement in MAS is generally bilateral, unilateral macrorchidism has been reported as an early and isolated clinical manifestation [225]. The diagnosis may be elusive until adulthood [226].
MAS is due to postzygotic gain of function mutation at codon 201 in the guanine nucleotide binding protein α-subunit (GNAS1) gene that encodes the GS-α protein [227]. In males with MAS, this mutation shows a mosaic distribution, because generalized mutation is lethal in utero [228,229]. The anomaly is followed by inactivation of both LHR and FSHR [230]. It is interesting to note that steroids secreted by the testis induce this precocious puberty, specifically by an autonomous hyperfunction of Sertoli cells without evidence of Leydig cell activation. This secretion would produce an early maturation of the hypothalamic-pituitary-testicular axis, and subsequently, a true precocious puberty [231]. Maturation occurs in both seminiferous tubules and Leydig cells. Testicular microlithiasis is a frequent finding (62%) [232,233].
Although the above-described developmental pattern is the rule, cases with different hormonal or clinical patterns have been reported. Some have isolated Sertoli cell hyperfunction, while others have only Leydig cell hyperfunction. Macrorchidism due to autonomous Sertoli cell hyperfunction shows abnormally elevated serum levels of inhibin B and anti-Müllerian hormone (AMH) along with decreased FSH and testosterone. Therefore, a pubertal inhibin B serum level would suggest a Sertoli cell hyperfunction with negative feedback on FSH secretion. These testes show Sertoli cell hyperplasia, absence of maturation of both germ cells and Leydig cells, with the subsequent lack of steroidogenesis [234]. This peculiar condition has been related to the presence of the somatic mutation in GNAS1 gene in Sertoli cells [235], but not in Leydig cells. The different early embryologic origin of precursor cells that contribute to Sertoli cell and Leydig cell lineages might underlay the differential occurrence of the mutated GNAS1 gene. In other cases, clinical, biochemical, and histological data suggest an isolated Leydig cell hyperfunction without precocious activation of Sertoli cells; this condition leads to complete spermatogenesis in some tubules [236]. In these cases, autonomous testicular function and gonadotropin suppression can persist for a long time.
Therapy in MAS is symptomatic. Precocious puberty is treated with aromatase inhibitors which block the conversion of testosterone into estradiol. Hypophyseal surgery is used in some cases of fibrous dysplasia; and bromocryptine, cabergoline, and long-acting somatostatin analog are used to stop the GH action [237].
Another cause of precocious puberty is tuberous sclerosis. Both gonadotropin-dependent and gonadotropin-independent mechanisms have been involved [115,238].