
Abstract
Malignant mesothelioma is an uncommon tumor that usually arises in the pleural cavity of adults with a history of asbestos exposure. Less frequently, it appears in the peritoneum or other mesothelial surfaces. Deciduoid mesothelioma is a rare subtype that has been found at both sites. Of the 3 reported cases in children, 2 originated in the mesenterium and 1 in the pleura. We describe a 4th case of pediatric, malignant, deciduoid mesothelioma and a third case in the mesenteric cavity. The patient was an 8-year-old girl who presented with abdominal pain and fullness. Workup revealed extensive involvement of the abdomen by a serosa-based tumor. The clinical and pathologic findings are described, and the pertinent literature is reviewed.
INTRODUCTION
Mesothelioma is an insidious neoplasm that develops from mesothelial cells. About 80% of mesotheliomas originate in the pleural cavity. Other sites include the peritoneal cavity, the tunica vaginalis, and the pericardium [1]. The predominant cause is inhalational exposure to asbestos, reported in 60% to 70% of all cases of pleural mesothelioma, and to a lesser extent, although still substantial, 15% to 30% of cases are peritoneal mesothelioma [1]. The incidence of mesothelioma in the United States and other developed countries appears to have peaked in 2000 and is now declining secondary to the control of asbestos exposure. However, in resource-limited settings, where asbestos mining is poorly regulated and industrial and household use of asbestos-containing products still proliferates, incidence rates of malignant mesothelioma are expected to increase [2]. Radiation to supradiaphragmatic fields in the treatment of malignancies (Hodgkin lymphoma, non-Hodgkin lymphoma, testicular cancer) has also been linked to a statistically significant increase in the risk of mesothelioma, although these cases account for only a small fraction of the total diagnosed [3]. Genetic studies have associated mesothelioma with a germline mutation in BAP1, inactivating the BAP1 nuclear deubiquitinase protein. [4].
Estimates based on epidemiologic data suggest that only 2% to 5% of all cases of mesothelioma present in the first 2 decades of life [5,6]. Our review of the medical literature for the past 20 years yielded 34 cases of pediatric mesothelioma of any type at any anatomic site [5–16]. All the neoplasms were malignant. Eleven were located in the pleura [5,6,8,9,15,16] and 23 in the peritoneum [5,7,10,14,16]. Only 3 of the children had a possible history of exposure to asbestos (fathers who work in the industry) [10]. Two had known previous malignancies, but which were not been treated with radiation [10,13].
The aim of the present report was to describe an extremely rare case of a malignant deciduoid mesothelioma in the abdomen in a child.
CASE REPORT
Clinical data
An 8-year-old girl presented to our tertiary pediatric medical center with weakness, anorexia, weight loss, intermittent diarrhea, and abdominal pain. On physical examination, she was found to be underweight, with abdominal tenderness and fullness. Laboratory studies revealed low levels of albumin (2.8 g/dL) and high-density lipoprotein cholesterol (26 mg/dL) suggesting a nutritional deficit. Additional findings were low levels of hemoglobin (7.8 g/dL, microcytic, hypochromatic) and iron (10 mcq/dL) and high levels of transferrin (190 mg/dL). Computed tomography revealed a large pelvic mass, mostly in the Douglas pouch, compressing the rectosigmoid, in addition to an omental cake, a thick peritoneum with a nodular appearance, and calcifications throughout the abdomen, along with a hypodense focus between the liver and ribs.
Treatment consisted of intravenous nourishment for 2 weeks to improve the patient's nutritional state followed by cytoreductive surgery with intraoperative, intraperitoneal, hyperthermic chemotherapy with cisplatinum at 41°C for 90 minutes. The surgical procedure included an extensive and detailed search for, and removal of, every macroscopically evident tumor mass in the abdominal and pelvic regions. The patient was then admitted to the intensive care unit, where she received continuous intraperitoneal chemotherapy for 4 days. Her general condition improved, and after she recovered from surgery, 6 courses of additional chemotherapy (pemetrexed, cisplatin) were administered, 3 weeks apart. Follow-up consisted of evaluation by magnetic resonance imaging and positron-emission tomography scans every 3 months. At present, approximately 24 months after surgery and 18 months after completion of treatment, she is free of disease and is doing well clinically.
Pathology
Cytologic examination of the peritoneal fluid showed clusters of malignant cells with a ball-like pattern. The cells were large, with wide, vacuolated cytoplasm and were seen on a background of neutrophils, lymphocytes, and macrophages (Fig. 1).
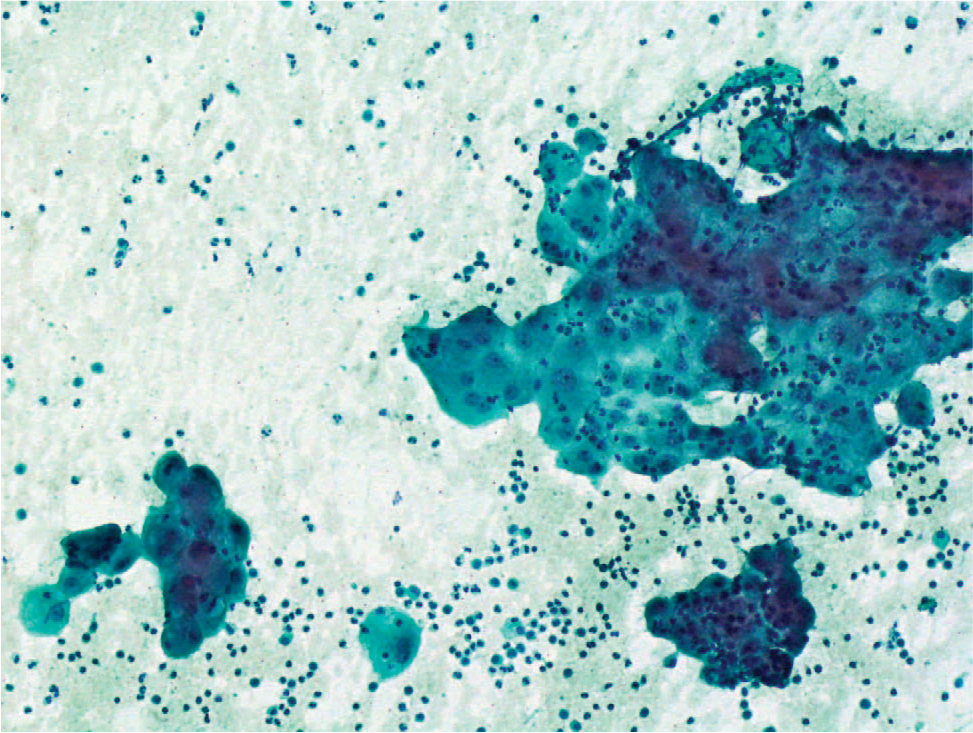
Cytologic test result showing clusters of malignant cells with a ball-like pattern. The tumor cells are large, with a wide, vacuolated cytoplasm on a background of neutrophils, lymphocytes and macrophages. (Papanicolaou stain, original magnification ×200). A color version of this figure is available online.
Numerous tumor specimens were obtained during cytoreductive surgery, from the peritoneum, diaphragm, falciform ligament, omentum, liver, stomach serosa, the surface of both ovaries and the right fallopian tube, the inferior vena cava, and external surfaces of the appendix and spleen. Macroscopically, the tumor consisted of grey-to-brown, nonuniform tissue with areas of necrosis. Microscopic study revealed papillary, tubular, and solid patterns formed by large cells with abundant, eosinophilic, partially foamy cytoplasm with a deciduoid appearance. The nuclei were large and vesicular, with prominent nucleoli (Fig. 2). Psammoma bodies were present throughout the tumor. On immunohistochemical studies, the tumor was diffusely positive for keratin 7, vimentin, and calretinin and was focally positive for CA 125 (Fig. 3). Staining for WT1 was negative. Staining for Ki-67 was positive in about 15% of the tumor cells. The microscopic and immunohistochemical findings were consistent with deciduoid, malignant mesothelioma.
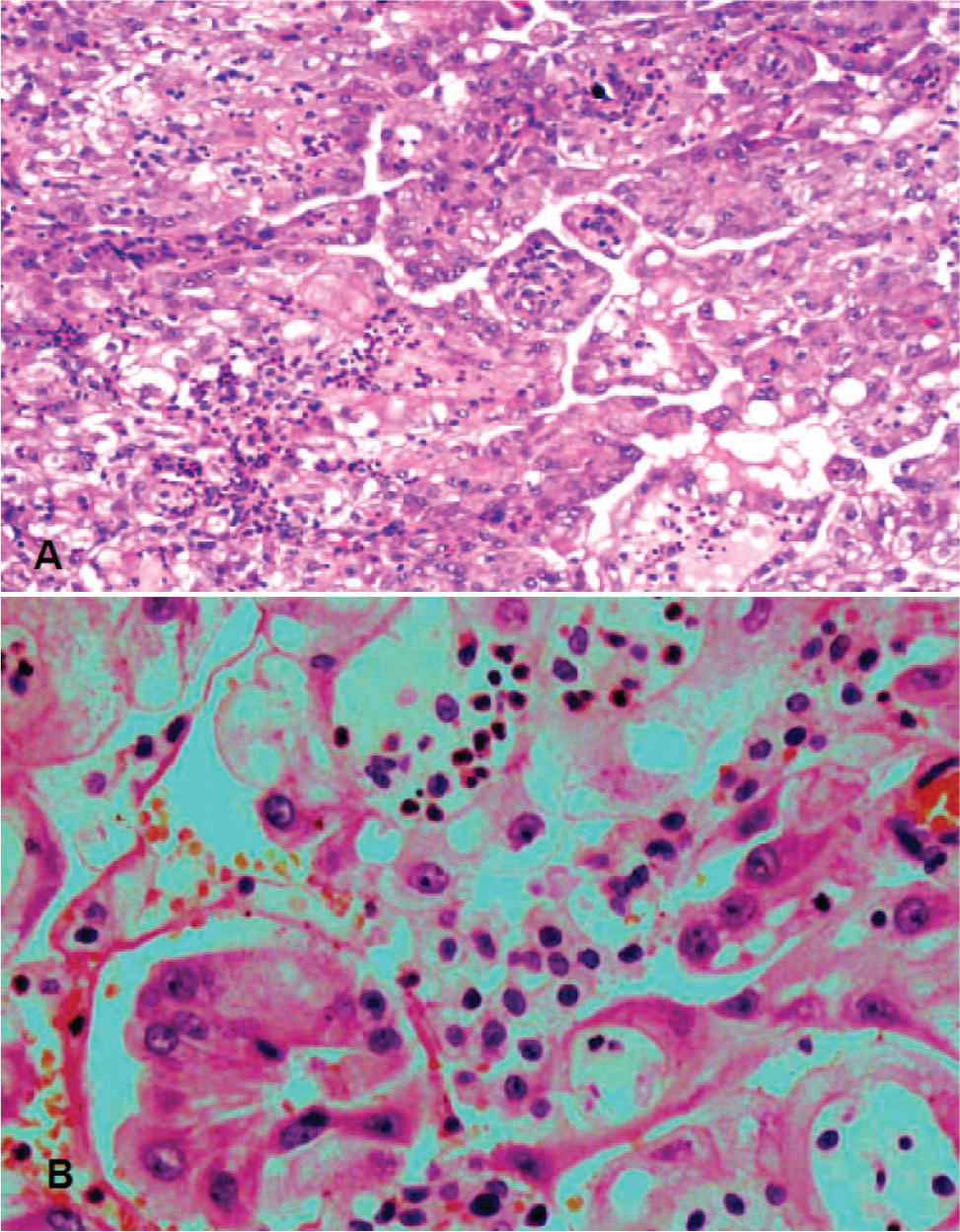
Histologic section showing the formation of large, deciduoid tumor cells in both papilla and solid areas. (
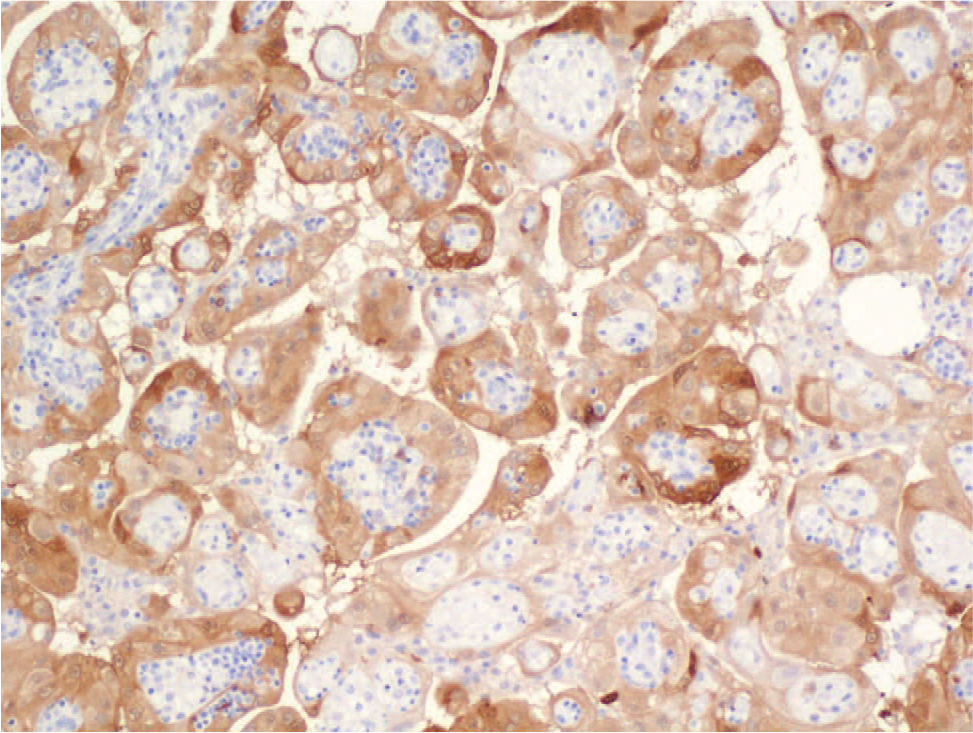
Histologic section stained with antibodies directed against calretinin showing cytoplasmic positivity of variable intensity in the tumor cells (calretinin immunostain, original magnification ×100). A color version of this figure is available online.
DISCUSSION
The term deciduoid mesothelioma was introduced by Nascimento and colleagues [17] in 1994 to describe a variant of epithelioid mesothelioma identified in 2 women aged 23 and 24 years with peritoneal neoplasms. This variant is a subtype of the classical epithelioid mesothelioma. The name reflects the morphologic resemblance of the variant to decidua or decidual-type changes of the female tract. The tumor mainly forms sheets of large, polygonal cells with abundant eosinophilic cytoplasm and vesicular nuclei. In 2002, Shia and colleagues [18] estimated that only 2% to 5% of all mesotheliomas are deciduoid. The rarity of these tumors was further highlighted in the review by Ordonez [16], which included 650 cases of adult mesothelioma evaluated by the Department of Pathology at the University of Texas; of which, 21 were found to be deciduoid. The diagnosis is even rarer in children, accounting for only 3 of the 34 pediatric cases of mesothelioma reported in the medical literature to date [16]. The remainder were regular epithelioid subtype (n = 16) [5,11,16], fibrous subtype (n = 2) [5], or biphasic with both epithelioid and spindle components (n 5 1) [11]; in 12 cases, no subtype was specified [6–8,10,12–15].
Interestingly, Ordonez [16] included the 1985 report of Talerman and colleagues [19], describing a 13-year-old girl with what the authors diagnosed as diffuse pseudotumoral deciduosis. However, the tumor was histopathologically similar to deciduoid mesothelioma, consisting of sheets of large cells with an eosinophilic and foamy cytoplasm, as observed in the present patient as well. Thus, the report of Talerman and colleagues [19] may be the first description of a deciduoid mesothelioma in a child.
The differential diagnosis of this subtype of mesothelioma includes benign-reactive, deciduosis carcinoma and metastatic carcinoma. In deciduoid-malignant mesothelioma, despite the low nuclear-to-cytoplasm ratio, the cells are larger than reactive mesothelial cells; contain coarse, granular chromatin; and have more-conspicuous nucleoli [20]. Deciduoid mesothelioma can be distinguished from metastatic carcinoma using epithelial membrane antigen immunostaining, which reveals a distinctive, brush borderlike pattern (“thick membrane”) in mesothelial cells that is not observed in adenocarcinoma [20]. In addition, ultrastructural studies may show microvilli that are circumferentially arranged around the neoplastic mesothelial cells, infiltrating stromal connective tissue, and interdigitation with stromal collagen fibers; these features, too, are absent in metastatic carcinoma [20]. These 2 differential diagnoses would be more appropriately considered in the adult population. In general, in children, this morphologic pattern is unusual and is seen only in other rare tumors. In the differential diagnosis of the present case, we considered tumors that could be compatible with the histologic findings, such as melanoma, epithelioid angiosarcoma, and poorly differentiated carcinoma. All were ruled out by immunohistochemical studies, which supported the diagnosis of mesothelioma.
The pleura are the most prevalent site of deciduoid mesothelioma. Of the 64 such tumors reported so far, to our knowledge, 39 arose in the pleura [16–18]. Fourteen of those patients were female and 25 were male, with ages ranging from 23 to 78 years, with the exception of one boy, who was 13 years old [16]. Another 23 tumors were located in the peritoneum. Sixteen of those patients were female and 7 were male; ages ranged from to 23 and 78 years, with the exception of 2 girls aged 13 and 15 years [16]. The remaining tumors were found in the testicles or pericardium. The present report adds another case of peritoneal deciduoid mesothelioma in a child to the literature.
Regarding etiology, previous exposure to asbestos was documented in 33 of the 61 cases of adult deciduoid mesothelioma but in none of the pediatric cases [16–18]. In addition, none of the children was subject to secondhand exposure, unlike some of the children with other subtypes of mesothelioma, whose parents had been exposed to asbestos in their work environment [10]. In our patient, no primary or secondary exposure to asbestos was recorded.
The favorable clinical behavior so far in this case may be attributed to the extensive debulking of the tumor during cytoreductive surgery and the administration of intraoperative and subsequent chemotherapy, as well as the noninvasive nature of the tumor, which seems to coat, rather than invade, parenchymal organs, and its relatively low proliferative rate.
In summary, to our knowledge, we report the 3rd case of a malignant, peritoneal deciduoid mesothelioma in a child. Raising awareness of such rare diseases and their clinical and pathologic characteristics is important to avoid possible underdiagnoses.