
Abstract
Diabetic embryopathy refers to a constellation of congenital malformations arising in the setting of poorly controlled maternal diabetes mellitus. Cardiac abnormalities are the most frequently observed findings, with a 5-fold risk over normal pregnancies. Although a diverse spectrum of cardiac defects has been documented, cardiac noncompaction morphology has not been associated with this syndrome. In this report, we describe a novel case of biventricular cardiac noncompaction in a neonate of a diabetic mother. The patient was a late preterm female with right anotia, caudal dysgenesis, multiple cardiac septal and aortic arch defects, and biventricular cardiac noncompaction. Examination of both ventricles demonstrated spongy myocardium with increased myocardial trabeculation greater than 50% left ventricular thickness and greater than 75% right ventricular thickness, with hypoplasia of the bilateral papillary muscles, consistent with noncompaction morphology. Review of the literature highlights the importance of gene expression and epigenomic regulation in cardiac embryogenesis.
INTRODUCTION
During early embryogenesis, fetal myocardium is composed of a trabecular network of spongy myocardium separated by deep recesses that allows blood to supply the developing myocardium. During weeks 3–8 of embryonic development, the ventricular myocardium progressively loses its immature, noncompacted trabeculae along and across decreasing gradients from the epicardium to the endocardium, from the base to the apex, and from left to right [1]. Cardiac noncompaction refers to the abnormal embryogenesis of the endocardium and myocardium, resulting in the lack of compaction of the early myocardial network [2]. Noncompaction was first described in 1932 from an autopsy performed on a newborn infant with aortic atresia/coronary-ventricular fistula [3]. The first case of isolated left ventricular cardiac noncompaction (LVNC) was published in 1984 by Engberding and Bender [2]. Noncompaction morphology is characterized by ventricular myocardium with a spongiform appearance. It is defined by the absence of well-formed left ventricular papillary muscles with myocardial trabeculation occupying at least 50% of the total thickness with hypoplasia of the left ventricular papillary muscles. Right ventricular (RV) involvement is defined by trabeculation occupying at least 75% of the total RV thickness [4]. Currently, the American Heart Association classifies isolated LVNC as a primary genetic cardiomyopathy [5]. Cardiac noncompaction morphology has also been implicated in a number of clinical syndromes including Barth syndrome [6], DiGeorge syndrome [7], myopathic changes with inclusions in skeletal muscle fibers [7], and Melnick-Needles syndrome [8]. The noncompaction phenotype, including the aforementioned syndromes, is usually limited to the left ventricle, although rare cases of RV or biventricular involvement have been documented [9–11].
In this case report, we describe the autopsy findings of a late preterm female neonate with biventricular cardiac noncompaction as well as features consistent with diabetic embryopathy (DE). Diabetic embryopathy is a syndrome characterized by a constellation of congenital abnormalities, including cardiac defects, arising in the setting of poorly controlled maternal diabetes. Although a diverse range of cardiac malformations has been documented in DE, cardiac noncompaction morphology has not been associated with this syndrome. In this report, unifying pathogenetic mechanisms of cardiovascular malformations arising within in a diabetic environment are discussed.
CASE REPORT
The patient was a late preterm female with prenatally diagnosed congenital heart disease, born at 36 weeks and 4 days to a 28-year-old G3P0111 woman with a pregnancy complicated by polyhydramnios and pregestational diabetes. No record of the mother's glucose control was documented during the current pregnancy secondary to poor antenatal care, but HbA1c at time of delivery was 7.3 g/dL (normal range <5.7 g/dL). The echocardiogram after birth demonstrated multiple cardiac defects including a complete atrioventricular (AV) septal defect, apical muscular ventricular septal defect (VSD), hypoplastic aortic valve and arch, and large bidirectional patent ductus arteriosus (PDA) (Fig. 1). Magnetic resonance imaging of the spine showed absent sacrum and squaring of the conus medullaris, consistent with caudal dysgenesis. In addition, magnetic resonance angiography showed no intra-abdominal inferior vena cava (IVC) and hepatic veins that drained directly to the heart. Renal ultrasound was negative for any anomalies. Cytogenetic studies showed a 46XX karyotype. Microarray-based comparative genomic hybridization demonstrated 39.2-megabase-long contiguous stretches of homozygosity on partial stretches of chromosomes 4, 7, and 11, suggesting consanguinity.
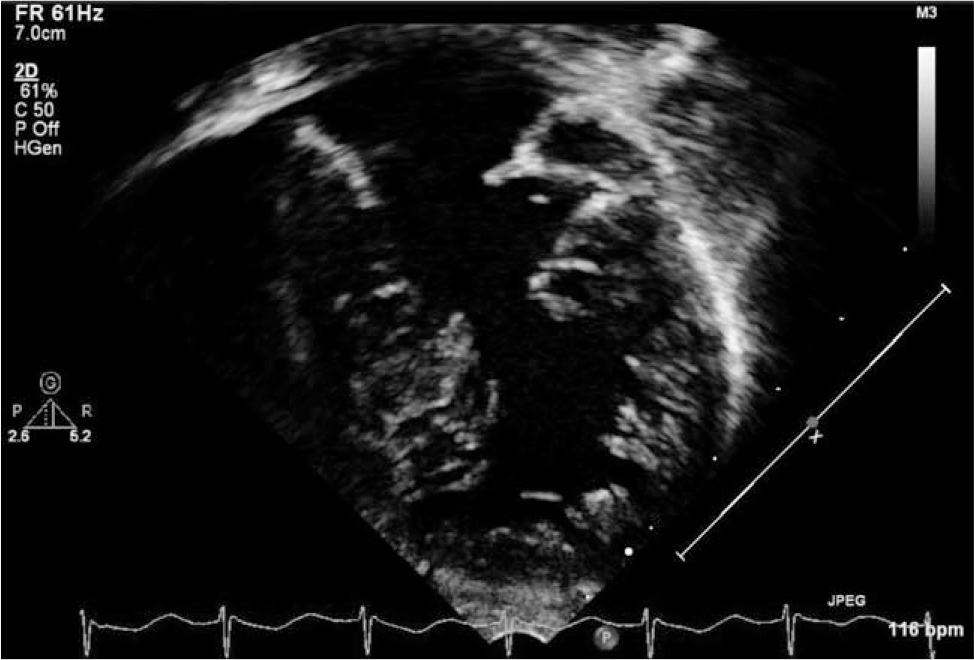
Transthoracic echocardiogram on day of life 0. Apical 4-chamber view of the left ventricle demonstrating relatively noncompacted left ventricular apical muscle, complete atrioventricular canal, and an apical muscular ventricular septal defect. The right ventricle is not shown.
Following birth, the patient developed respiratory distress requiring intubation with high flow oxygen at 3 days of life and later developed Escherichia coli pneumonia. The clinical course was also significant for progressive heart failure with severe AV regurgitation. The patient was ultimately transitioned to comfort care and passed away on day of life 17 (corrected calculated gestational age 39 weeks).
At autopsy, the external exam showed an average for gestational age female neonate with complete absence of the external and internal right ear (right anotia). The internal examination showed a constellation of findings consistent with DE. Multiple cardiac and aortic arch anomalies were noted, consistent with her aforementioned radioimaging studies. In addition, she had interruption of the IVC with azygous continuation and an absent sacrum. The placenta was not available for pathologic review.
On gross examination, the heart demonstrated massive cardiomegaly and biventricular hypertrophy with prominent spongiform changes and hypoplasia of the papillary muscles bilaterally (Fig. 2), as well as abnormal cardiac shunts, including a complete AV septal defect and apical muscular VSD. The IVC was noted to be interrupted at the hepatic portion, with continuation of the azygous vein that drained directly into the left atrium. Two hepatic veins were identified. One hepatic vein drained into the right atrium and the other into the left atrium. The bilateral atrial appendages phenotypically resembled that of the normal finger-shaped left atrial appendage, although the bilateral atria showed normal right and left internal morphologies. Additionally, a hypoplastic aortic arch was present with narrowing of the isthmus between the left carotid artery and large PDA. The aortic valve was mildly stenotic with 3 hypoplastic cusps. Microscopic sections of myocardium by routine hematoxylin and eosin and Masson-Trichrome stains showed increased trabeculations of the left ventricle greater than 50% of the total thickness and right ventricle greater than 75% of total thickness, consistent with bilateral ventricular cardiac noncompaction morphology (Fig. 3).
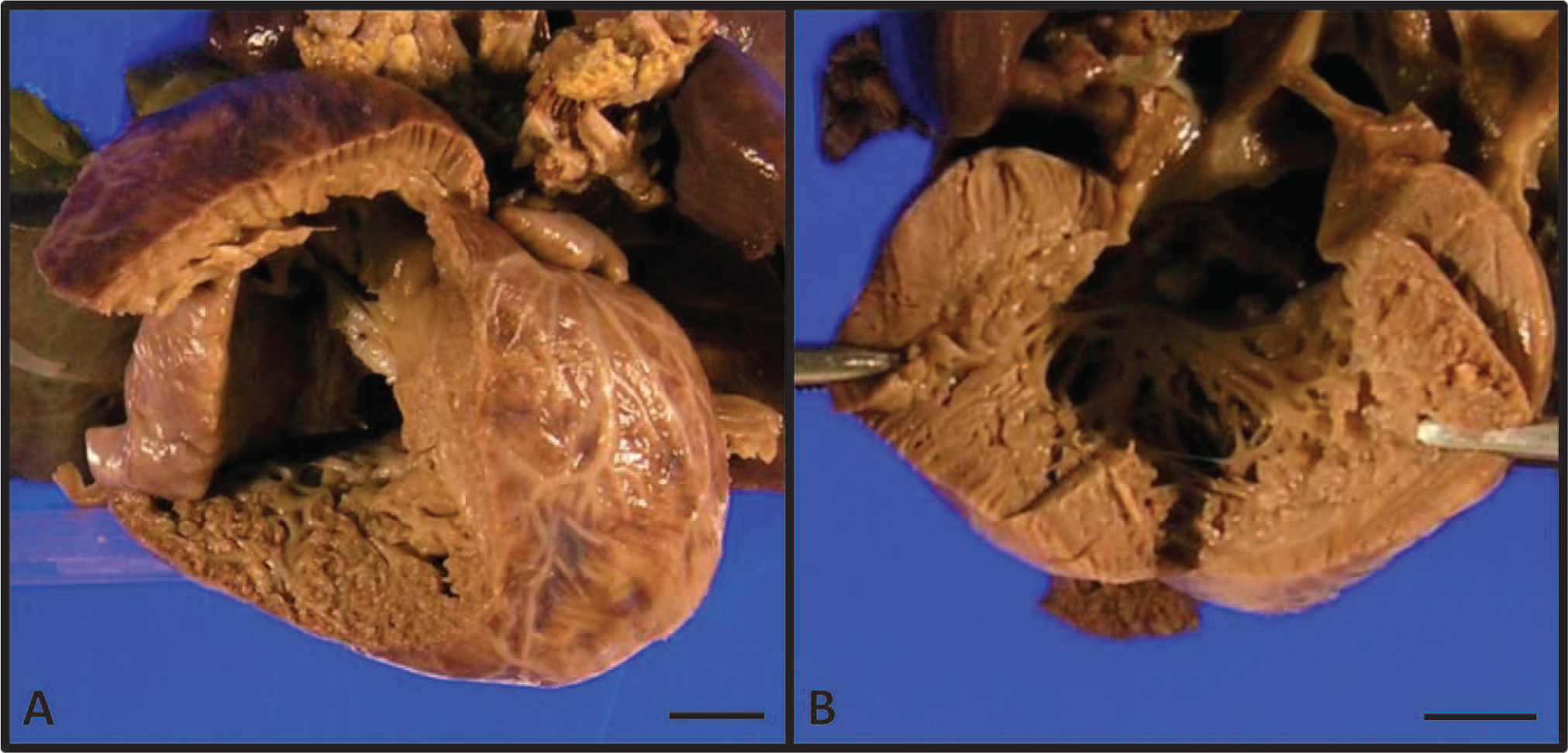
Gross examination of the right (
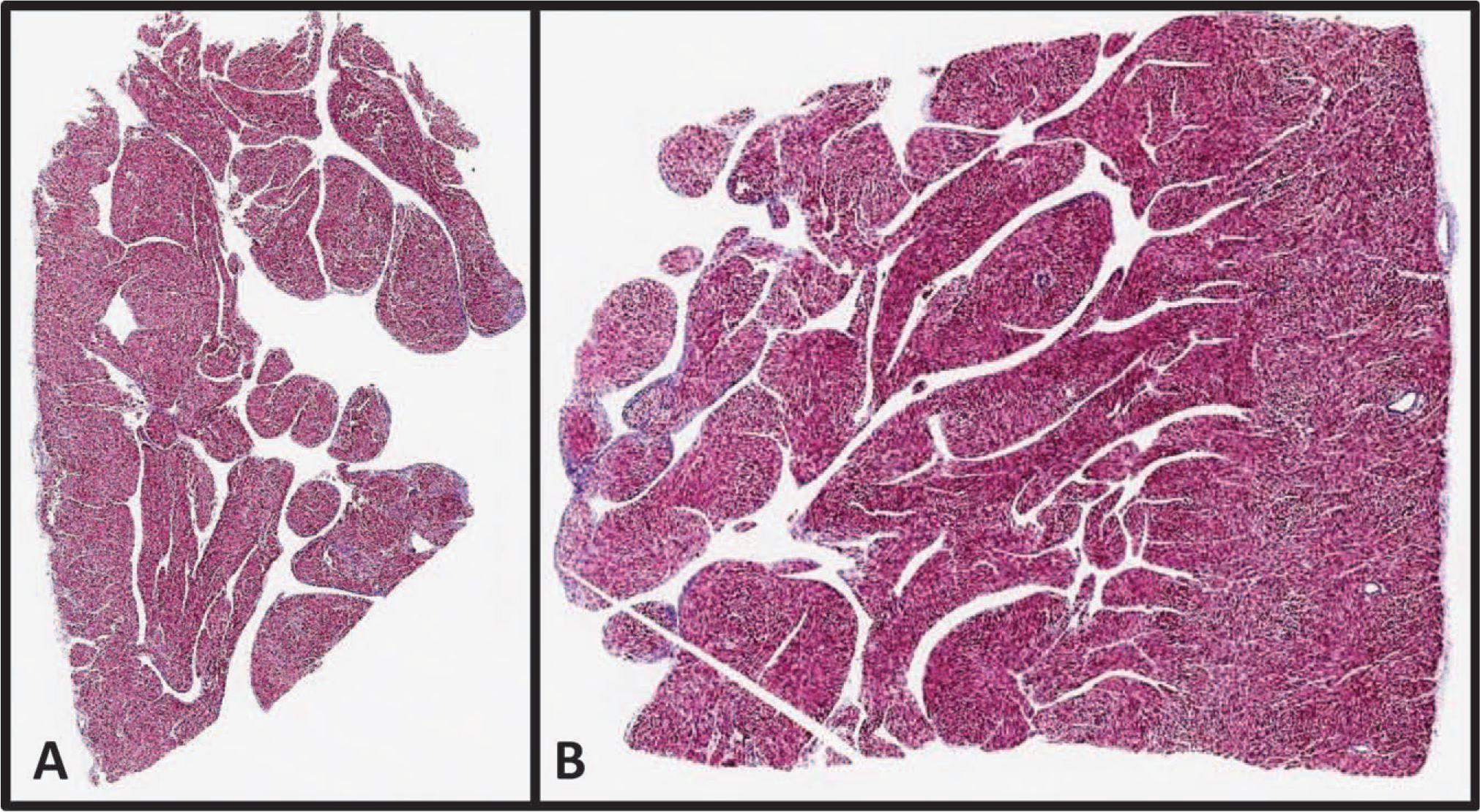
Microscopic examination of the right (
DISCUSSION
Diabetic embryopathy refers to a diverse host of congenital malformations and can be seen in up to 10% of all diabetic pregnancies [12]. The most common congenital malformations include cardiovascular defects and central nervous system defects (neural tube defects and caudal dysgenesis); both are leading causes of mortality and morbidity in children of diabetic mothers [13–16]. Structural cardiac abnormalities in DE run a range of defects and occur in up to 5% of diabetic pregnancies, with a 5-fold risk over normal pregnancies [17]. Cardiac anomalies in DE can affect every compartment of the developing heart, resulting in heart tube dilation, abnormal heart looping, hypoplastic ventricular development, and outflow tract stenosis.
Abnormal embryogenesis in DE is attributed to the role of elevated glucose as a potent teratogen. For example, oxidative stress in the maternal diabetic environment appears to contribute to malformations of the outflow tracts of the developing heart by interrupting the migration of cardiac neural crest cells during the development of the endocardial cushions [18,19]. Cardiac neural crest defects in mouse models suggest that the downregulation of genes involved in the development of the cardiac neural crest (Bmp4 [20–22], Msx1 [23–25], and Pax3 [26]) could contribute to the pathogenesis of maternal diabetes–induced congenital heart defects [27]. Factors involved in the transforming growth factor β (TGF-β) signaling pathway, a pathway that plays a role in heart development regulation, have also been implicated and are downregulated by maternal diabetes [28].
The pathogenesis for cardiac noncompaction has been linked to several gene mutations and deletions. Isolated forms of LVNC have been linked to familial forms. The causative gene mutation identified in this setting is seen in the X-linked inherited gene G4.5 (encoding for tafazzin) [29]. Other gene mutations or deletions seen in association with cardiac noncompaction include the α-dystrobrevin (DTNA) gene [30,31], Z-band alternatively spliced PDZ-motif protein (ZASP) [32,33], FKBP12 [34], the lamin A/C (LMNA)–related sequence gene [35], NKX2.5 [31], TBX5 [31], cardiac-specific gene (CSX) [36], human cardiac sodium channel α-subunit gene (SCN5A) [37], and various sarcomere protein genes (α-cardiac actin [ACTC], cardiac troponin T [TNNT2], and β-myosin heavy chain [MTH7]) [38], among others that are detailed in a recent review by Udeoji and colleagues [39]. A recent genome-wide linkage analysis of a family with autosomal dominant LVNC demonstrates that the locus for autosomal dominant LVNC maps to a 6.8-megabase region on human chromosome 11p15 [38].
Although mechanisms for cardiac malformations are diverse and multifactorial, common pathways for abnormal cardiac development/compaction in the maternal diabetic setting are highlighted by several studies. For example, common mechanisms for cardiac malformations in DE and cardiac noncompaction are described in TGF-β signaling studies, such as Smad7, a negative regulator in the TGF-β signaling pathway [40]. The TBF-β pathway plays a major role in cell proliferation, differentiation, apoptosis, and embryonic development, and its downregulation has been implicated in DE [28]. In Smad7 mouse models, the majority of Smad7 mutant mice demonstrated multiple cardiac defects in cardiovascular development, including VSD and outflow tract malformation as well as the noncompaction phenotype. Noncompaction in Smad7-deficient mice was thought to be secondary to abnormal ventricular compaction. In addition to Smad7, FKBP12 is another negative regulator for TGF-β signaling, and also has also been implicated in the cardiac noncompaction phenotype [34].
Epigenetic changes, including chromatin remodeling and histone modification studies, are also implicated as pathogenic factors in both DE and cardiac noncompaction. Because infants of diabetic mothers present with variable phenotypic presentations, one possible explanation for this variability is the differential role of epigenetic modifiers [41]. In hyperglycemic environments, excess glucose can lead to a high availability of acetyl-coenzyme A (acetyl-CoA) cell nuclei, which acts as a direct enzymatic substrate for histone acetylation [42]. Moreover, the enzyme responsible for the generation of acetyl-CoA, adenosine triphosphate citrate lyase, has been shown to play a critical role in embryogenesis, with targeted deletions resulting in embryonic demise [43]. This suggests that an intricate balance of epigenetic factors is crucial in normal embryonic development. In cardiac noncompaction, histone demethylases have been shown to play a role in the noncompaction phenotype. The histone demethylases Jarid2/Jumonji and Jmjd6/Ptdsr have been shown to contribute to normal cardiac outflow tract septation by silencing gene expression. Jarid2-null mice have been shown to demonstrate myocardial noncompaction, VSDs, and double-outlet right ventricle [44,45], whereas Jmjd6-deficient mice show VSDs, pulmonary artery hypoplasia, and double-outlet right ventricle [46].
Emerging evidence shows that epigenetic mechanisms can alter mitochondrial DNA. Specifically, type 2 diabetes and obesity have been implicated as triggers of epigenetic alterations in the mitochondrial genome. In diabetic subjects, the genes PGC1α and Tfam (regulators of mitochondrial biogenesis), as well as the genes of mitochondrial oxidative metabolism (COX7A1 and NDUFB6), appear to be repressed by increased DNA methylation [47]. This is of particular interest as mitochondrial DNA mutations have been implicated in cases of left ventricular noncompaction [48].
CONCLUSION
In summary, we report a novel case of biventricular cardiac noncompaction arising in the setting of DE. Although mechanisms for cardiac development are complex and multifaceted, underlying abnormalities in gene expression and aberrant epigenetic mechanisms may play a role in cardiovascular malformations within the diabetic environment.
Footnotes
ACKNOWLEDGMENTS
We would like to acknowledge the family for their consent and the UCLA Department of Pediatrics and Cardiology for the clinical workup of this patient.