
Abstract
We present a case of pure erythroleukemia, diagnosed at autopsy, in a dysmorphic premature infant who died of multiorgan failure within 24 hours of birth. Dysmorphic features included facial and limb abnormalities with long philtrum, microagnathia, downturned mouth, short neck as well as abnormal and missing nails, missing distal phalanx from the second toe, and overlapping toes. Internal findings included gross hepatomegaly and patchy hemorrhages in the liver, splenomegaly, and cardiomegaly; and subdural, intracerebral, and intraventricular hemorrhages. Histology revealed infiltration of bone marrow, kidney, heart, liver, adrenal, lung, spleen, pancreas, thyroid, testis, thymus, and placenta by pure erythroleukemia. Only 6 cases of congenital erythroleukemia have been previously reported with autopsy findings similar to those of this case. The dysmorphic features, although not fitting any specific syndrome, make this case unique. Congenital erythroleukemia and possible syndromes suggested by the dysmorphic features are discussed.
INTRODUCTION
Congenital leukemias originate in utero, and symptoms are present at birth or within the first 4 weeks of life [1,2]. Symptoms vary, with hepatosplenomegaly and skin lesions being especially frequent. Infiltration of other locations (eg, lymph nodes, testis, kidneys) are more unusual [1]. Diagnosis of congenital leukemia requires proliferation of immature white blood cells, involvement of nonhematopoietic tissues, and absence of any disease that could mimic congenital leukemia, such as infections [1,2]. The incidence of congenital leukemia ranges from 1 to 5 per 1 million live births and represents 0.8% of all childhood cases of acute leukemia [3]. However, the risk of developing childhood cancer such as leukemia has been shown to be linked to malformation syndromes with, for example, Down syndrome, carrying a markedly increased incidence for acute leukemias [3–5].
Acute myeloid leukemia (56%–64%) and acute lymphoblastic leukemia (21%–38%) make up the majority of neonatal leukemias [3]. Erythroleukemia is extremely rare in the neonatal period, with only 6 previously reported cases [1,6–10]. Acute erythroid leukemia is characterized by a predominantly (⩾50%) erythroid population in the bone marrow. According to the World Health Organization classification of 2008, acute erythroid leukemia is divided into 2 subtypes: erythroid/myeloid leukemia, with ⩾50% erythroid precursors and ⩾20% myeloid blasts of nonerythroid population; and pure erythroleukemia, with ⩾80% erythroblasts. Erythroleukemia is recognized by the proliferation of bizarre, multinucleated, megaloblastoid erythroblasts with expression of glycophorin A and CD71 and negative staining for CD3, CD4, CD6, CD8, CD10, and CD20 [2]. We report a unique case of congenital erythroleukemia in a premature neonate with dysmorphic features.
CASE REPORT
A male neonate weighing 1425 g was born to a primigravid mother at 32+2 weeks of gestation by emergency cesarean section.
The pregnancy had been uneventful until 29+4 weeks of gestation when ultrasonography showed borderline reduced amniotic fluid index. Two subsequent scans showed hepatomegaly and cardiomegaly. An emergency section was done due to fetal compromise. The baby had APGAR values of 2 at 1 minute, 4 at 5 minutes, 7 at 10 minutes, and 8 at 20 minutes. He was given oxygen and treated by intubation and ventilation.
He continued to present with significant ventilatory requirements requiring high-flow oxygen and had cardiomyopathy, hypotension, and profoundly low hemoglobin requiring blood transfusions, fluid boluses, dopamine, dobutamine, noradrenaline, milrinone, and hydrocortisone. At 18 hours of age, he went into cardiac arrest and was resuscitated and recovered but continued to have episodes of intermittent bradycardia and hypotension. In addition, cranial ultrasonography showed possible infarct in the cerebellum. He also continued to be anemic, with low platelets and a coagulopathy. He had hyperkalemia, hypercalcemia, and hyperphosphotemia from birth, with worsening metabolic acidosis and intractable lactate levels. This, combined with poor cardiac function, gave a very poor prognosis. With parents' consent, a decision was made to stop intensive care, and he died within 24 hours of delivery.
Autopsy findings
Autopsy of the 1-day-old male neonate born at 32+/40 weeks of gestation showed biometric measurements smaller than expected for the stated gestation (foot length of 55 mm, which corresponds to 29 weeks of gestation). He exhibited dysmorphic facial features consisting of a long philtrum, microagnathia, downturned corners of the mouth, and short neck. He had external abnormalities of pallor, petechial hemorrhages in the sclerae and chest wall, jaundice, abnormal and missing nails on hands and feet, missing distal phalanx from the second toe, and overlapping toes (Fig. 1). There was gross hepatomegaly (weight, 211 g; expected weight, 62.5 ± 30 g) and patchy hemorrhages of liver, splenomegaly (weight, 7.83 g; expected weight, 4.7 ± 5.4 g), cardiomegaly (weight, 18.9 g; expected weight, 10.1 ± 4.4 g), and subdural, intracerebral, and intraventricular hemorrhages. Lungs showed congestion and patchy hemorrhages.
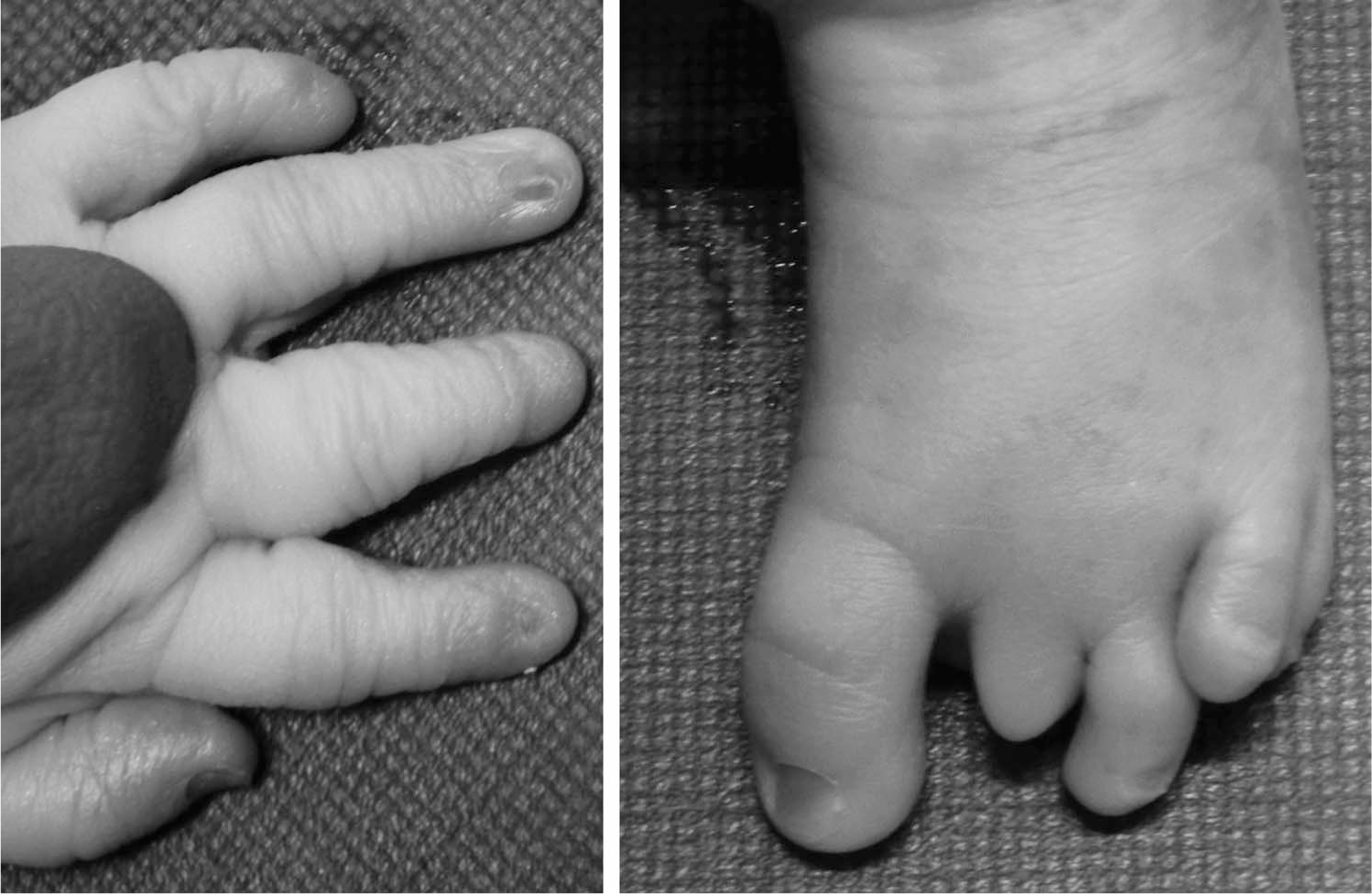
Macroscopic view of the patient's hand with abnormal and missing nails and foot with missing distal phalanx from the second toe and overlapping toes.
Histological examination of bone marrow, kidney, heart, liver, adrenals, lung, spleen, pancreas, thyroid, testis, thymus, and placenta showed infiltration by immature cells that had the morphology of blasts with very round hyperchromatic nuclei and minimal cytoplasm (Figs. 2 and 3). Immunohistochemistry for CD117 highlighted the immature cells. Most expressed glycophorin A and CD71, confirming erythroid differentiation (Fig. 2 and 3), with occasional cells expressing E-cadherin. Few cells expressed MPO (granulocyte/myelocyte differentiation) and occasional cells expressed CD61 and von Willebrand factor (megakaryocyte differentiation). The appearances and immunophenotypes were of pure erythroleukemia with most tumour cells showing mature stage differentiation and only some early/immature cells. No fresh tissue was taken for polymerase chain reaction analysis because the diagnosis was not suspected at the time of autopsy.
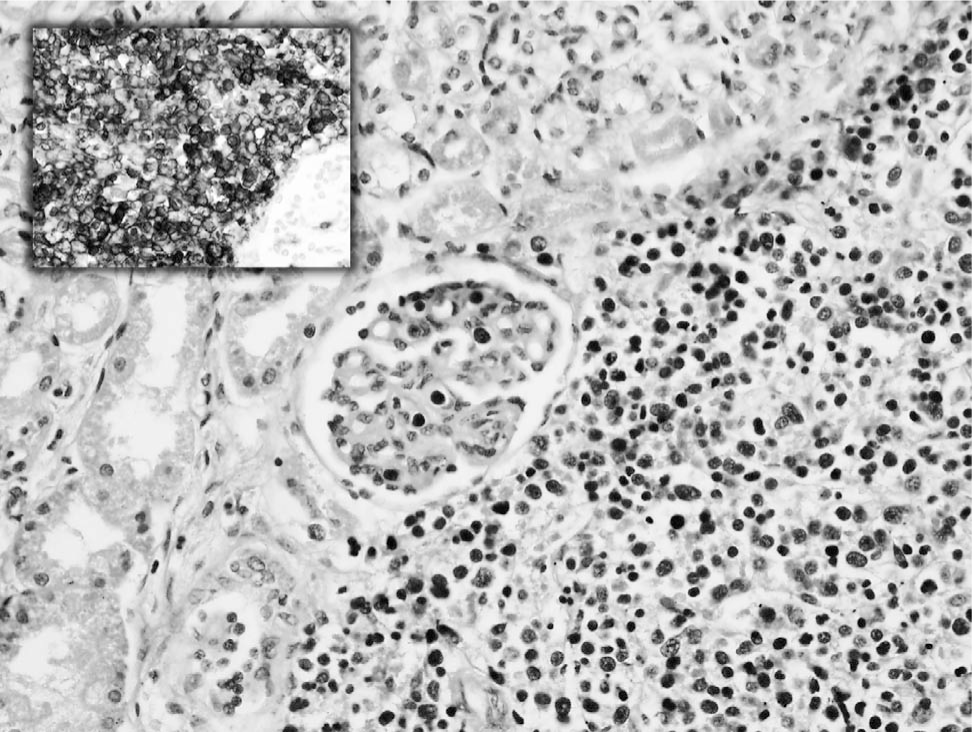
Kidney with glomeruli, tubules, and parenchyma infiltrated by immature cells that had the morphology of blasts with very round hyperchromatic nuclei and minimal cytoplasm (hematoxylin and eosin, x20). Inset, kidney stained with glycophorin A, highlighting the erythroid differentiation of the blast cells diffusely infiltrating the parenchyma (glycophorin A, x40).
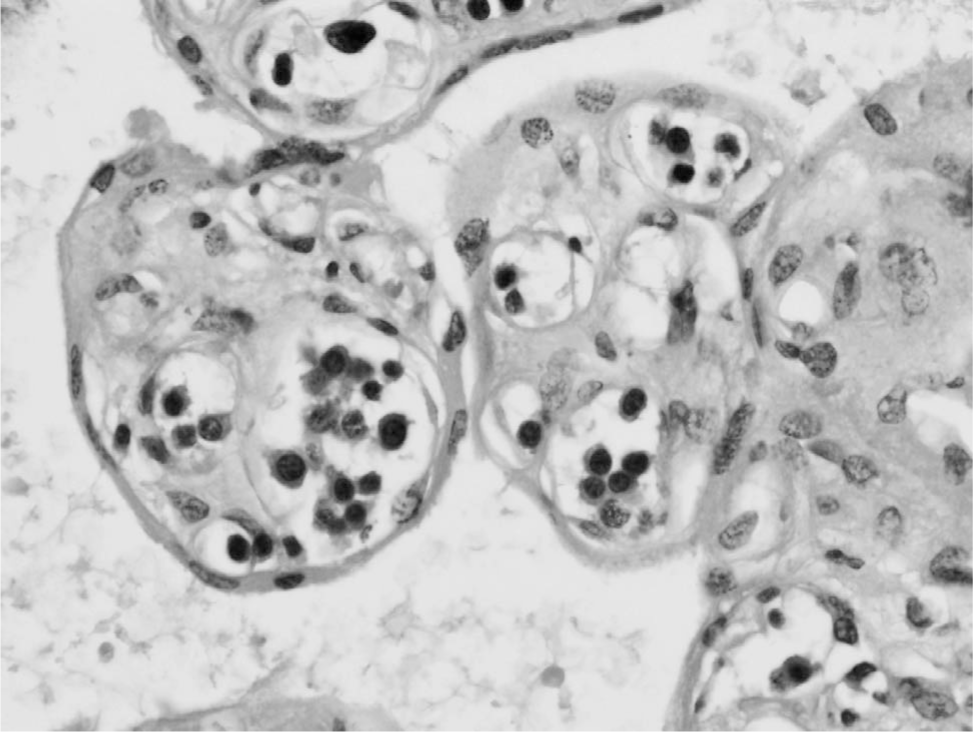
Placenta containing villi, within which are fetal vessels filled with immature cells that had morphology of blasts with very round hyperchromatic nuclei and minimal cytoplasm (hematoxylin and eosin, x40).
In addition, hemorrhagic necrosis of the liver, hyaline membranes, pulmonary hemorrhages, and ischemic enteritis; and subdural, intradural, intracerebral, and intraventricular hemorrhages were noted on histology.
Metabolic tests showed no abnormalities. Bacteriologic and virologic finding were negative. Array comparative genomic hybridization using 60000 probes across genome showed a normal male sex chromosome complement with no abnormalities.
DISCUSSION
This rare case of congenital erythroleukemia is unusual because of its aggressive postnatal clinical course and association with dysmorphic features without detectable genetic abnormality.
The major part of the clinical picture and aggressive course observed was explained by the underlying congenital erythroleukemia, with death resulting from failing marrow and leukemic infiltration of most organs and tissues by erythroid blast cells. Most of the autopsy findings of massive infiltration of nonhematopoietic tissues with blasts were in keeping with those seen in other cases of congenital erythroleukemia [1,6–10]. Indeed, the observed hepatosplenomegaly is the most common clinical feature of leukemia in neonates, occurring in approximately 80% of patients [1,3,8]. Unlike most reported cases of congenital erythroleukemia, diagnosis of congenital leukemia was not suspected premortem because no blasts were noted in blood films due to numerous transfusions with blood products, and bone marrow was not examined.
Most of the previously reported cases of congenital erythroleukemia have exhibited a more indolent clinical course, although the majority resulted in mortality within 1 week to 2 months of delivery [1,6–10]. One published case reported a comparably aggressive clinical course leading to death within the first 3 days [6]. Interestingly, that case resembled the current one in many ways, including the baby being delivered prematurely due to fetal distress, severe asphyxia, and brain hemorrhages [6]. Similar to the current case, no blasts were noted in blood tests, but on autopsy, most of the organs were infiltrated by leukemic cells. However, unlike the current case, there was no hepatosplenomegaly, liver being relatively spared [6]. These 2 cases may perhaps be a more aggressive subset of erythroleukemia already adversely affecting the fetus in utero [6].
In adults, pure erythroleukemia often has prior diagnosis of a myelodysplastic syndrome, acute erythroid leukemia of erythroid/myeloid type, or myeloproliferative neoplasm and is frequently therapy related [11]. The karyotype is almost always abnormal and highly complex. It has a highly aggressive clinical behavior [11]. Congenital erythroleukemia shares many morphological and immunohistochemical features with the adult version but deserves to be regarded separately in view of the difference in presentation.
The occurrence of leukemia with the accompanying dysmorphic features raised the possibility of an underlying malformation syndrome. Indeed, it is well documented that Down syndrome in particular carries a markedly increased incidence of acute leukemias, including erythroblastic leukemia [3,5]. Due to the presence of leukemia, limb anomalies and dysmorphic features with normal array comparative genomic hybridization Noonan/RASopathies spectrum, Rubinstein-Taybi, Adams-Oliver, and WT syndromes in particular were considered. However, craniofacial features were not those seen in RASopathies [12]. By contrast, although some of the craniofacial features such as micrognathia were in keeping with Rubinstein-Taybi syndrome, the digit abnormalities were felt not to be in concordance with this diagnosis [13]. Similarities between the terminal limb defects and those in AdamsOliver syndrome, where abnormal nails and short or missing fingers and toes can be seen, were evident, but the lack of typical scalp or underlying bone defects made that diagnosis unlikely [14].
Leukemia together with terminal limb defects raised the possibility of WT syndrome as this is characterized by ulnar and radial defects such as deviated, bifid, or hypoplastic thumbs, clinodactyly, and cutaneous syndactyly together with anemia, pancytopenia, leukemia, and lymphoma [15]. It can occur at various ages, and there is a range of clinical presentations, even between affected individuals from the same family [15]. The underlying genetic mechanism is not yet known [15]. Therefore, although the clinical findings could be suggestive of WT syndrome, the lack of positive family history and absence of possible molecular testing make this diagnosis difficult to apply.
In cases of leukemia it is important to recognize the presence of underlying syndromes, such as Down syndrome, as it has been shown that treatment may need to be adjusted in particular situations [5]. However, it is impossible to extrapolate the effect of a possible syndrome on the clinical course seen here, particularly because a similarly aggressive clinical course has been noted in the absence of additional dysmorphic features and limb anomalies [6]. Indeed, most of the previously reported cases of congenital erythroleukemia have led to death [1,6–8]. In fact, in this vulnerable group, even for the more common congenital acute lymphoid and myeloid leukemias, the overall survival rates have been reported as 17% and 25%, with high relapse rates of 73% and 50%, respectively [3].
In conclusion, we present a unique case of aggressive congenital erythroleukemia in a premature infant with dysmorphic features which did not fit any specific syndrome. To the best of our knowledge, a case like this has not been described before.
Footnotes
ACKNOWLEDGMENT
We thank Dr Hammad Khan, consultant neonatologist at St Thomas' Hospital.