
Abstract
Perineuronal nets (PNNs) are specialized extracellular matrix structures that enwrap select neuronal populations—particularly, parvalbumin-positive inhibitory interneurons. Under physiological conditions, they stabilize synaptic connectivity, regulate excitatory– inhibitory balance, and contribute to the closure of developmental critical periods. Rather than being static scaffolds, PNNs dynamically regulate neuronal excitability, synaptic plasticity, and circuit maturation across the lifespan. Notably, accumulating evidence indicates that the disruption of these functions represents a cross-diagnostic mechanism underlying shared circuit vulnerabilities across multiple brain disorders. In autism spectrum disorder, schizophrenia, bipolar disorder, epilepsy, Alzheimer’s disease, amyotrophic lateral sclerosis, and substance use disorders, alterations in PNN density, composition, and remodeling are consistently associated with disrupted PV interneuron function, impaired network oscillations, and aberrant circuit stability. A central paradox thus emerges: excessive PNN stabilization may prematurely restrict plasticity during development, whereas pathological degradation or dysregulation in adulthood destabilizes mature circuits and increases vulnerability to excitotoxicity, neuroinflammation, and maladaptive memory persistence. These context-dependent effects highlight the translational potential of targeting PNNs to restore circuit balance. Together, these findings position PNNs as critical regulators of the balance between stability and adaptability in neural circuits and underscore their potential as translational targets for circuit-level therapeutic interventions.
Keywords
1 Background
Perineuronal nets (PNNs) have emerged as a convergent biological substrate across neurodevelopmental, neuropsychiatric, and neurodegenerative disorders. Rather than representing a disease-specific epiphenomena, growing evidence indicates that PNN dysfunction reflects a cross-diagnostic mechanism that perturbs shared neural circuit endpoints, including the excitation–inhibition balance, synaptic stability, and experience-dependent plasticity. A central paradox underlies this process: PNNs confer structural and functional stability that is necessary for late developmental maturation and circuit consolidation, whereas excessive stabilization or impaired remodeling in adulthood may constrain adaptive plasticity. This imbalance between stability and flexibility is increasingly recognized as a unifying pathological principle across diverse disorders.
The extracellular matrix (ECM) of the central nervous system (CNS) is a highly specialized structural and biochemical network that extends far beyond a passive scaffold. Composed of glycoproteins, proteoglycans, glycosaminoglycans, and structural proteins such as collagen and elastin, the ECM maintains neural tissue architecture while actively shaping cellular behaviors and signaling cascades [1, 2]. Within the CNS, the ECM assumes unique properties from other organ systems, reflecting the functional demands of neuronal networks. Rather than remaining static, ECM components are continuously synthesized, remodeled, and degraded throughout development, learning, and responses to injury, thus underscoring the dynamic role of the neural ECM in regulating synaptic connectivity, neuronal excitability, axonal guidance, and neuroimmune interactions [3, 4].
In early neurodevelopment, ECM remodeling occurs continuously in response to intrinsic genetic programs and extrinsic environmental cues [5, 6]. Proteolytic enzymes, including matrix metalloproteinases (MMPs), a disintegrin and metalloproteinase with thrombospondin motifs, and hyaluronidases, regulate ECM turnover and enable circuit plasticity during sensitive periods of synaptogenesis and refinement [7]. The transient restructuring of ECM components during these windows facilitates the elimination of inappropriate synaptic connections while stabilizing essential ones. During adulthood, ECM turnover slows considerably but never ceases. Learning, environmental enrichment, and stress can induce localized ECM remodeling, thereby altering synaptic strength and functional connectivity, whereas pathological conditions such as traumatic brain injury, ischemia, neuroinflammation, and neurodegeneration often accelerate ECM degradation or aberrant deposition, leading to circuit dysfunction [8–10].
Several ECM constituents are important for the formation and function of PNNs, which represent a specialized subdomain of the neural ECM. Chondroitin sulfate proteoglycans (CSPGs), including aggrecan, brevican, neurocan, and versican, form the structural backbone of PNNs. These proteoglycans anchor to hyaluronan chains, synthesized by hyaluronan synthases, and are stabilized by link proteins such as cartilage link protein 1 [11]. Tenascin-R, a neural glycoprotein, cross-links CSPGs and contributes to the dense lattice-like structure characteristic of mature PNNs [11, 12]. Additional molecules such as laminin, fibronectin, and reelin participate in axonal guidance, synaptic adhesion, and the modulation of glutamate receptor trafficking [13]. Importantly, individual ECM molecules exert distinct and sometimes opposing influences on synaptic plasticity. Laminin and tenascin-R tend to promote long-term potentiation, thereby enhancing memory consolidation, whereas heparin-binding growth-associated molecule can inhibit long-term potentiation under certain conditions [10, 14, 15]. The balance among these molecular signals is therefore critical for maintaining healthy neural network dynamics.
Although often discussed as discrete structures, PNNs do not exist in isolation. Core PNN components, including CSPGs, tenascin-R, and hyaluronan-associated link proteins, are shared with the diffuse ECM, forming a continuum rather than a strict boundary between PNN- associated and non-PNN matrix domains [4, 11]. Consequently, changes in PNN density or composition frequently coincide with broader ECM reorganization, particularly during heightened neural activity, neuroinflammation, or injury. Activity-dependent ECM remodeling, mediated by protease release, glial signaling, and altered ECM synthesis, can therefore reshape both PNNs and the surrounding matrix, biasing circuits toward stability or plasticity depending on context [1]. This integrated ECM perspective helps to explain why similar PNN alterations can produce distinct functional outcomes across brain regions and disease states.
One of the most important roles of the ECM, and especially PNNs, is the regulation of the excitatory–inhibitory (E/I) balance. PNNs preferentially enwrap parvalbumin (PV)-expressing inhibitory interneurons, where they modulate synaptic inputs and firing precision [6, 16]. Through this regulation, PNNs indirectly control gamma oscillations and other network rhythms that are essential for cognition, working memory, and sensory processing [17]. The disruption of ECM structure via genetic mutation, immune activation, or enzymatic degradation can tilt this balance toward hyperexcitability or hypoexcitability, thus contributing to the pathophysiology of diseases such as epilepsy, schizophrenia, and autism spectrum disorders (ASD) [4].
Aberrant ECM remodeling has emerged as a common pathway across neurodevelopmental, neurodegenerative, and psychiatric disorders. In neurodevelopmental conditions, premature PNN formation can prematurely close critical periods of plasticity, leading to maladaptive circuit stabilization [18]. By contrast, excessive ECM degradation in adulthood can destabilize circuits, impair memory retention, and increase vulnerability to excitotoxic or oxidative damage [19]. Whether triggered by systemic infection, autoimmune disease, or neurodegenerative pathology, chronic neuroinflammation often accelerates ECM breakdown via microglial activation and protease secretion [20]. Furthermore, ECM alterations can impair the regenerative capacity of the brain after injury by creating a non-permissive environment for axonal regrowth—a well-documented phenomenon in spinal cord injury and multiple sclerosis models [21].
Given its central role in maintaining neural homeostasis and plasticity, the ECM—and PNNs in particular—has emerged as both a biomarker and therapeutic target across a broad spectrum of CNS disorders. Advances in in vivo imaging, proteomics, and transcriptomics now enable the study of ECM and PNN dynamics in living human brains, supporting translational efforts to modulate these structures for therapeutic benefit [22].
2 PNN Physiology
PNNs are highly organized, lattice-like ECM structures that primarily surround fast-spiking, PV-expressing γ-aminobutyric acid-ergic interneurons, though they also enwrap some excitatory neurons [6, 11]. Structurally, PNNs consist of a hyaluronan backbone, CSPGs, link proteins, and crosslinking glycoproteins such as tenascin-R. PNN assembly is a tightly regulated, multistage process that coincides with critical developmental windows. Together, this creates a microenvironment that regulates ion diffusion, neurotransmitter signaling, and protease activity in the neuronal perisynaptic space [11]. By acting as selective diffusion barriers, PNNs stabilize synaptic contacts and maintain the ionic conditions necessary for optimal neuronal firing and signal transmission [16, 17].
During postnatal development, PNNs gradually emerge around PV interneurons in parallel with the closure of critical periods of sensory and cognitive plasticity [6, 23]. This temporal alignment suggests that PNNs play a central role in “locking in” functional circuitry once optimal wiring is formed. In the visual cortex, for example, PNN formation coincides with ocular dominance plasticity closure, a well-studied model of developmental critical periods [24].
Once established, PNNs serve multiple overlapping functions. A primary role is synaptic stabilization, in which PNNs restrict the lateral mobility of α-amino-3-hydroxy-5-methyl-4- isoxazolepropionic acid and N-methyl-d-aspartic acid receptors at synapses, thereby stabilizing excitatory transmission [16]. PNNs also provide neuroprotection by limiting oxidative stress and buffering ionic gradients; this protects neurons from excitotoxic insults [25]. In parallel, PNNs regulate plasticity by stabilizing mature synapses while restricting the formation of new synaptic connections, effectively acting as molecular “brakes” on circuit remodeling [4]. Finally, PNNs contribute to homeostatic control by regulating the perisynaptic environment, including protease activity and neurotransmitter diffusion [26] (Figure 1).
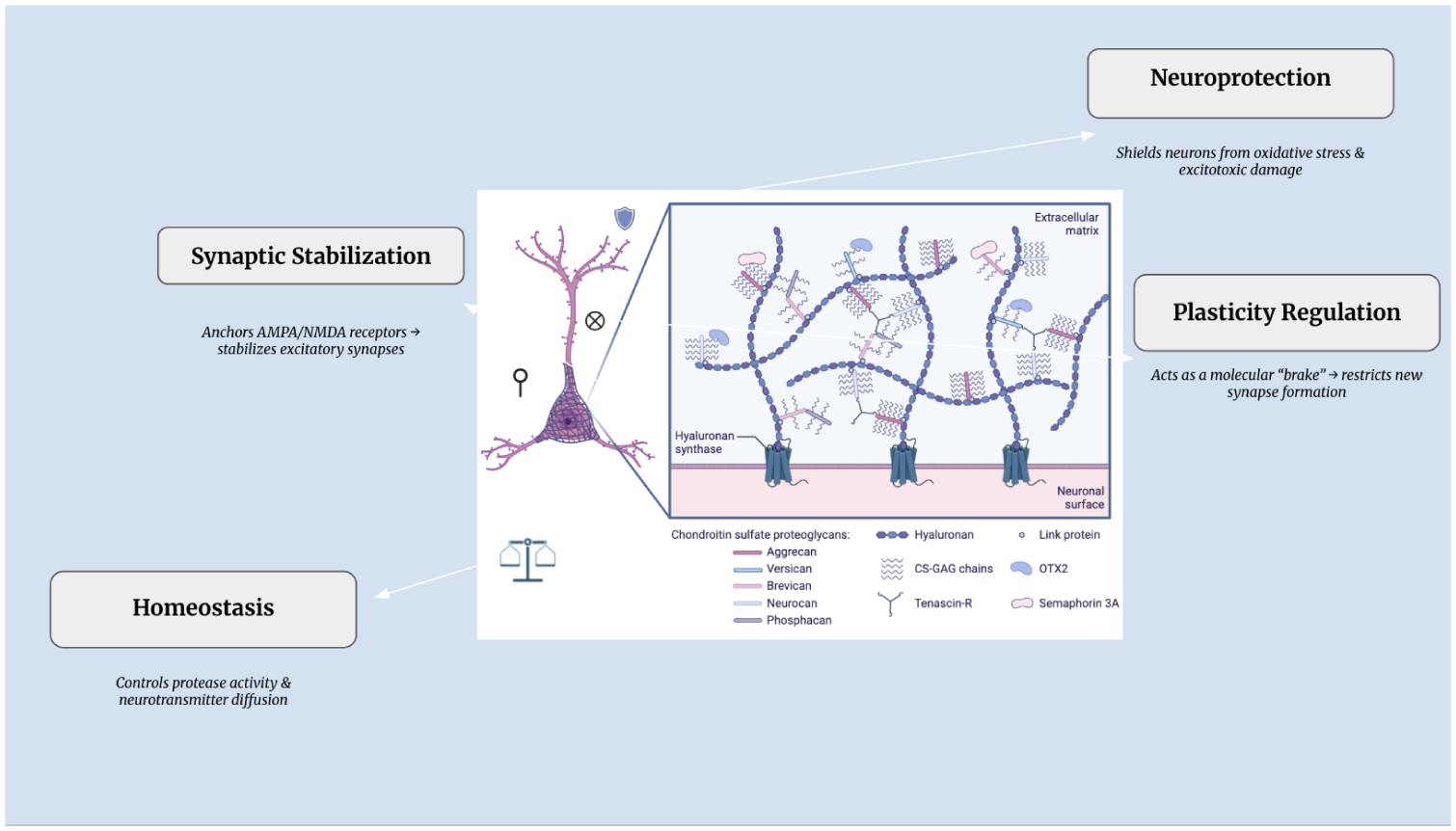
Roles of Perineuronal Nets in Synaptic Function. Created in BioRender. Sikder, S. (2026) https://BioRender.com/v7stoi4
Some of these functions continue in adults; PNNs continue to protect neurons from oxidative damage and excitotoxic stress while constraining aberrant synaptic remodeling that might destabilize established networks. However, this stability comes at the cost of reduced neuroplasticity, which limits learning-induced synaptic modifications and regeneration in the mature brain [16].
3 PNNs in Psychiatric and Neurological Disorders
3.1 ASD
3.1.1 Pathological Features
PNN abnormalities are increasingly implicated in ASD. Postmortem human studies indicate reduced PNN density around PV neurons in the globus pallidus (a basal ganglia structure involved in motor control and social behavior), whereas cerebellar Purkinje cells (important for sensorimotor integration) exhibit preserved PNNs [27] (Table 1). These findings suggest that PNN pathology in ASD is circuit-selective rather than globally distributed, thus potentially contributing to heterogeneous behavioral phenotypes.
Summary of Perineuronal Net Changes in Autism Spectrum Disorder Models
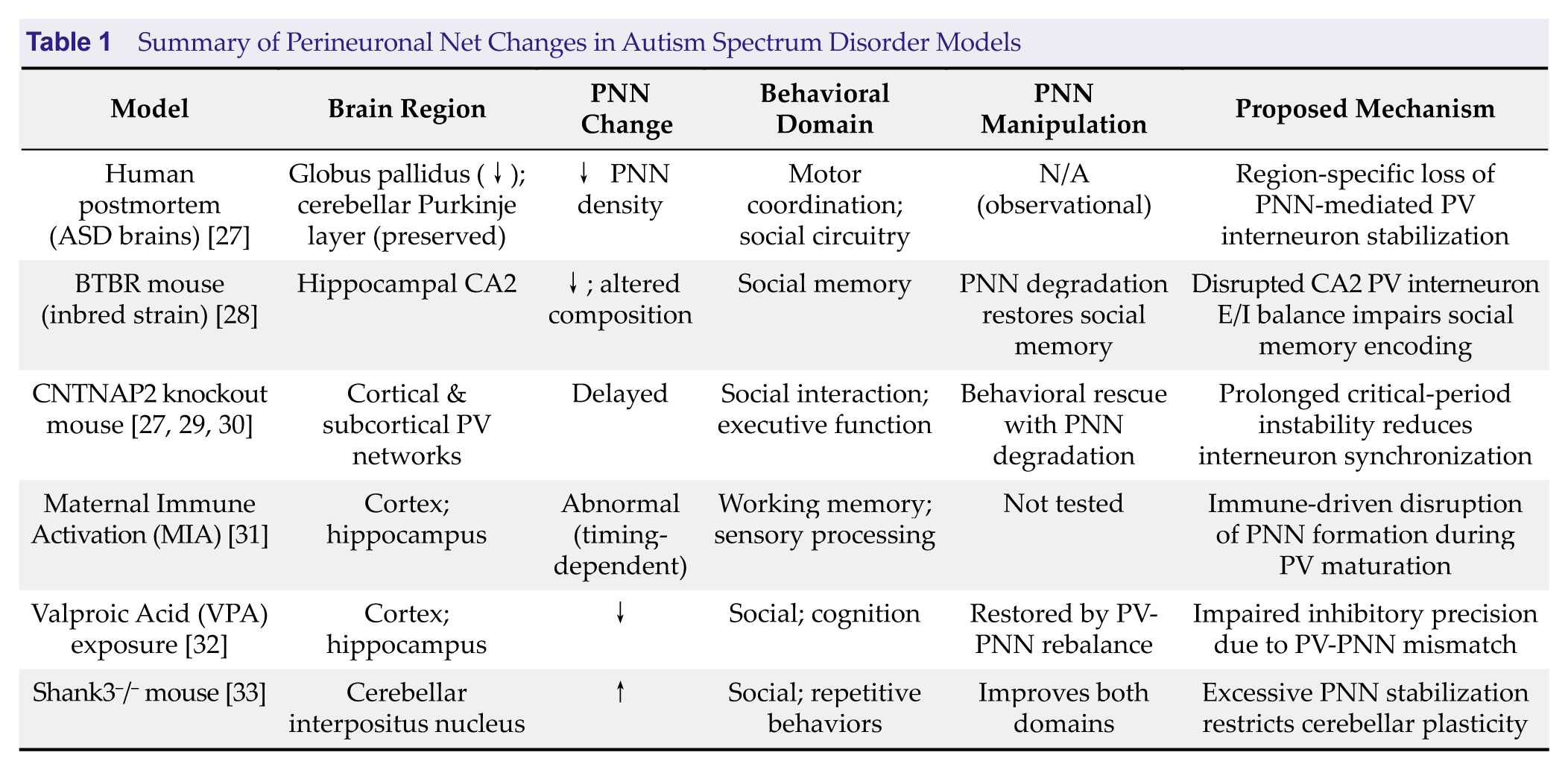
Abbreviations: ASD, Autism spectrum disorder; PNN, Perineuronal net; PV, Parvalbumin.
3.1.2 Evidence from Experimental Models
Evidence from multiple ASD-relevant models further supports a role for disrupted PNN regulation in circuit dysfunction. In the BTBR mouse model, atypical PNNs in the hippocampal CA2 region impair social memory, with enzymatic degradation restoring social recognition but not repetitive behaviors, thereby indicating circuit-specific contributions of PNNs to ASD phenotypes [28].
More broadly, models such as CNTNAP2 mutations and environmental perturbations (including maternal immune activation and prenatal valproic acid exposure) demonstrate that disrupted timing of PNN maturation impairs inhibitory circuit development, E/I balance, and experience-dependent plasticity [29–32]. By contrast, other models highlight excessive PNN stabilization as a constraint on plasticity; increased PNN intensity in the cerebellum of Shank3 knockout mice limits circuit flexibility, and targeted degradation rescues behavioral deficits [30]. Similarly, increased hippocampal PNNs in Nf1+/- mice are associated with reduced ECM remodeling capacity, further supporting dysregulated PNN turnover as a contributing factor [31].
Collectively, these findings highlight aberrant PNN development as a convergent pathway through which genetic mutations and environmental risk factors disrupt inhibitory circuit maturation and experience-dependent plasticity in ASD. Importantly, both insufficient and excessive PNN stabilization can produce ASD-relevant behavioral phenotypes, thus underscoring the context- dependent role of PNNs in circuit regulation. By linking molecular ECM alterations to circuit-level dysfunction, PNN abnormalities provide a unifying mechanistic framework that integrates heterogeneous ASD etiologies while identifying circuit-specific opportunities for therapeutic intervention.
3.2 Neurodegenerative Diseases
3.2.1 Alzheimer’s Disease
A hallmark feature of Alzheimer’s disease is the loss of PNN integrity, particularly around PV interneurons. This is driven partly by microglial activation and ECM protease release and occurs independently of classic amyloid plaque deposition [32, 33]. Under physiological conditions, intact PNNs stabilize synaptic contacts, buffer oxidative stress, and constrain tau aggregation and neurofibrillary tangle formation [34]. PNN degradation in Alzheimer’s disease disrupts inhibitory circuit function, reduces PV interneuron excitability, and shifts the E/I balance toward hyperexcitability. These circuit-level changes are correlated with cognitive impairment and memory deficits in human patients and animal models. Mechanistically, activated microglia directly promote PNN breakdown through protease secretion, thereby linking neuroinflammation to ECM destabilization. Notably, the experimental depletion of microglia preserves PNN integrity even in the presence of amyloid pathology, underscoring immune-mediated ECM remodeling as a primary driver of PNN loss rather than a secondary consequence of plaque accumulation [34]. Together, these findings suggest that maintaining PNN integrity may stabilize inhibitory networks, protect neurons from tau pathology, and potentially slow Alzheimer’s disease progression.
3.2.2 Amyotrophic Lateral Sclerosis
In amyotrophic lateral sclerosis (ALS), disruption of PNN integrity also contributes to selective neuronal vulnerability. The SOD1-G93A transgenic mouse, a widely used ALS model, recapitulates key pathological features of the disease, including progressive motor neuron degeneration, muscle weakness, and reduced lifespan. In this model, PNN degradation occurs around spinal motor neurons and closely correlates with disease severity and neuronal loss. PNN loss reduces the structural and metabolic support of motor neurons, in part through alterations of CSPGs and other ECM components. This increases their susceptibility to excitotoxicity and oxidative stress and accelerates motor neuron degeneration [35]. Experimental interventions, such as mesenchymal stromal cell transplantation, demonstrate partial PNN restoration and associated functional improvements such as motor outcomes, supporting the idea that preserving or rebuilding PNNs may enhance neuronal resilience [35].
Collectively, these findings indicate that the progressive dismantling of PNNs contributes to motor neuron instability in ALS. Targeting ECM remodeling and preserving PNN structure may therefore represent a complementary therapeutic strategy for slowing disease progression and protecting vulnerable motor circuits.
3.2.3 Parkinson’s Disease
Emerging evidence suggests that PNNs in basal ganglia circuits are altered in Parkinson’s disease, although human studies remain limited. Animal models exhibit PNN remodeling in the striatum and globus pallidus—key regions for motor control and procedural learning. These changes involve both reduced PNN density and aberrant molecular composition, disrupting E/I balance [36]. Such alterations impair synaptic plasticity, exacerbate dopaminergic dysregulation, and may contribute to the abnormal motor learning and progressive motor deficits that are characteristic of Parkinson’s disease. Beyond motor control, basal ganglia PNN changes are also likely to influence cognitive and psychiatric domains, including executive function, motivation, and mood regulation, highlighting the role of ECM integrity in supporting multiple behavioral processes.
3.2.4 Multiple Sclerosis and Traumatic Brain Injury
In multiple sclerosis and traumatic brain injury, ECM breakdown generates bioactive fragments, such as low molecular weight hyaluronan and remodeled CSPGs, that amplify neuroinflammation via microglial and astrocytic activation and pattern-recognition pathways. PNN disruption destabilizes inhibitory circuits and hinders oligodendrocyte progenitor maturation and remyelination [37]. In lesion and peri-lesion zones, CSPG accumulation and altered ECM composition create growth-inhibitory terrain that limits axonal regeneration and circuit repair [37]. Targeted enzymatic digestion of CSPGs with chondroitinase ABC can relieve this inhibition, promote axonal sprouting and synaptic remodeling, and support functional recovery, illustrating the context-dependent, dual roles of CSPGs/PNNs in either constraining aberrant activity or—when persistently dysregulated—blocking repair [38, 39].
3.2.5 Normal Aging
PNNs respond to external stressors, learning, and immune challenges, implicating them in normal aging and neurodegenerative progression [19–21]. During aging, chronic low-grade neuroinflammation activates microglia and astrocytes, which release MMPs and other proteolytic enzymes that accelerate ECM breakdown and destabilize PNNs [20, 21]. Acute immune challenges further increase cytokines such as interleukin-1β and tumor necrosis factor, thereby upregulating MMP activity, disrupting PNN structure, and impairing synaptic stability and plasticity [20]. CNS injuries or stressors that trigger reactive gliosis shift ECM composition toward inhibitory CSPGs, thereby interfering with neuronal communication and limiting regeneration [21].
Across neurodegenerative and neuro- developmental disorders, PNNs therefore function as double-edged swords: providing neuroprotection when intact but restricting repair once damaged. Interventions that restore PNN or ECM balance—through the enzymatic digestion of CSPGs, microglial activity modulation, or structural support of PNNs—show promise for rescuing behavioral and functional deficits. These findings suggest that therapeutic strategies must navigate the dual nature of PNNs, preserving their protective functions while enabling plasticity and regeneration.
3.3 Neuropsychiatric Disorders
3.3.1 Pathological Features
Abnormal PNN density and composition occur in major psychiatric disorders such as schizophrenia and bipolar disorder. Postmortem studies from schizophrenia patients have consistently revealed reduced PNNs within the prefrontal cortex, amygdala, and entorhinal cortex [41, 42]. These deficits are associated with impaired PV interneuron function and are correlated with psychotic symptom severity, supporting the hypothesis that disrupted interneuron maturation and synaptic plasticity drive disease pathogenesis [43]. Notably, PNN maturation in the prefrontal cortex continues into early adulthood (the typical onset period for schizophrenia), highlighting a critical developmental window during which aberrant PNN formation destabilizes inhibitory circuits [42]. Molecular alterations in CSPGs, reelin, and MMPs further disrupt PNN scaffolding, thus impairing the synaptic connectivity, plasticity, and network oscillations necessary for normal cognitive and emotional processing [43, 44]. Bipolar disorder exhibits similar but milder reductions in PNN density, particularly within dorsolateral prefrontal circuits, suggesting a shared mechanism of inhibitory circuit destabilization across mood and psychotic disorders [43]. These findings indicate that PNN abnormalities compromise the interneuron-mediated regulation of cortical networks, contributing to the cognitive, emotional, and perceptual disturbances characteristic of severe psychiatric conditions [41–43, 44].
3.3.2 Evidence from Experimental Models
In bipolar disorder, PNN reductions are milder than those in ASD but still occur in the prefrontal and limbic circuits, potentially impairing inhibitory control and emotional dysregulation [43]. Rodent models of major depressive disorder and post-traumatic stress disorder also show PNN alterations that destabilize PV interneurons and disrupt the E/I balance in cortical and hippocampal networks [44, 45]. These changes contribute to maladaptive memory persistence, heightened fear responses, and impaired cognitive flexibility, mirroring the behavioral phenotypes of these disorders. Preclinical studies suggest that targeted interventions, such as the enzymatic digestion of PNNs, pharmacologic modulation of ECM components, or activity-dependent remodeling through behavioral therapies, may restore circuit flexibility and alleviate cognitive and affective deficits. These findings highlight PNNs as key regulators of mood- and cognition-related network stability, with subtle alterations offering potential therapeutic targets.
3.3.3 Post-Traumatic Stress Disorder
Animal studies indicate that PNN modulation influences fear memory processes that are relevant to post-traumatic stress disorder. The genetic loss of key ECM components that form PNNs impairs long-term fear memory consolidation, highlighting PNNs as potential regulators of traumatic memory stability and targets for post-traumatic stress disorder interventions [46]. Intact PNNs protect consolidated fear memories from erasure, and their targeted disruption reopens plasticity, thus creating opportunities to therapeutically modify fear memories [47] Nonetheless, direct clinical evidence of PNN alterations in patients with post-traumatic stress disorder remains limited, and more work is needed to translate these mechanistic findings into human pathology and treatment strategies.
3.4 Epilepsy
3.4.1 Evidence from Experimental Models
In epilepsy, PNN proteolysis has been implicated in increased seizure susceptibility. Rodent models demonstrate that PNN enzymatic degradation, mediated by MMPs and other extracellular proteases, reduces the structural scaffolding of PV interneurons and weakens their inhibitory control over excitatory circuits [48, 49]. This loss of perisomatic inhibition destabilizes network oscillations, increasing hyperexcitability and the likelihood of seizure initiation and propagation. Pharmacological inhibition of ECM-degrading enzymes can mitigate seizure frequency and severity, highlighting the protective role of intact PNNs in maintaining the E/I balance [4, 48, 49].
The effects of PNN disruption are context- dependent. Although PNN loss may shorten tonic–clonic seizures, it can paradoxically increase myoclonic activity or spike-and-wave discharges, reflecting differential effects on local versus long-range network synchronization [48, 49]. Mechanistically, PNN disruption alters synaptic connectivity, decreases PV interneuron firing precision, and interferes with ion buffering and extracellular potassium regulation, all of which amplify network excitability [48, 49]. Furthermore, seizure activity itself can induce PNN proteolysis, creating a feedforward cycle that further destabilizes inhibitory circuits [48, 49].
3.4.2 Pathological Features
Emerging human evidence supports the translational relevance of PNNs in epilepsy. Postmortem studies of temporal lobe epilepsy patients have revealed region-specific changes in PNN density and ECM composition within hippocampal and cortical seizure foci, often accompanied by interneuron dysfunction and chronic hyperexcitability [50, 51]. PNN alterations appear to follow age- and disease-stage-dependent trajectories, reflecting the maladaptive remodeling of inhibitory circuits [51]. Although PNN loss commonly increases seizure susceptibility, excessive or aberrantly stabilized PNNs can create circuit rigidity and impair homeostatic plasticity after recurrent seizures. These findings position PNNs as context-sensitive regulators of circuit stability and highlight their potential as temporally constrained therapeutic targets.
3.5 Substance Use Disorders
3.5.1 Cocaine and Opioids
PNNs regulate addiction-related neuroplasticity by stabilizing drug-associated memories that drive relapse. Animal studies indicate that targeted PNN degradation in the medial prefrontal cortex or lateral hypothalamus disrupts the cue- and context-induced reinstatement of cocaine- seeking behavior without impairing natural reward processing, suggesting selective effects on maladaptive memory circuits [52, 53]. This suggests that PNNs help to maintain the synaptic scaffolding and E/I balance required for the persistence of drug-associated memories, particularly via PV interneuron interactions.
Developmental timing also influences vulnerability: adolescent methamphetamine exposure in rats alters PNN density and PV interneuron expression in the medial prefrontal cortex in a sex-dependent manner, highlighting critical windows for long-term circuit remodeling [54]. Similarly, binge alcohol exposure increases PNN intensity in the insula, and the enzymatic degradation of these PNNs modulates decision- making in ethanol preference tasks, indicating that PNNs contribute to the maintenance of compulsive or aversion-resistant alcohol-seeking behaviors [55, 56]. Opioids and cannabis also induce region- and drug-specific patterns of PNN remodeling, although these remain less studied [52–56].
In addiction, PNN function is context- dependent. Although PNN degradation can weaken drug-associated memories and facilitate behavioral flexibility during abstinence, the excessive or poorly timed disruption of PNNs may destabilize prefrontal inhibitory control circuits and increase vulnerability to compulsive drug seeking. Moreover, drug exposure itself can induce activity-dependent PNN remodeling via dopaminergic signaling, neuroinflammatory pathways, and microglial activation, raising the possibility that PNN changes reflect both adaptive responses to repeated drug exposure and contributors to long-term circuit pathology [52–56].
3.5.2 Environmental Factors
Environmental factors, including diet, exercise, and exposure to natural rewards, dynamically modulate PNN expression and composition in reward-related circuits such as the medial prefrontal cortex, nucleus accumbens, and lateral hypothalamus [53]. These activity-dependent changes in PNNs influence synaptic plasticity, PV interneuron function, and the E/I balance, affecting the persistence of drug-associated memories and relapse risk. Enriched environments or voluntary exercise enhance PNN formation and strengthen inhibitory control, potentially reducing compulsive drug-seeking behaviors, whereas high-fat or highly palatable diets may disrupt PNN integrity and promote maladaptive reward processing [53].
Overall, PNNs serve as critical molecular substrates for the encoding and persistence of addiction-related memories, stabilizing drug- associated neural circuits while integrating environmental influences such as diet, exercise, and natural rewards. By modulating PV interneuron function, synaptic plasticity, and the E/I balance within key reward circuits, PNNs represent promising targets for interventions aimed at weakening maladaptive memories, enhancing behavioral flexibility, and ultimately preventing relapse.
4 Conclusions and Future Directions
PNNs are central regulators of neuronal stability, synaptic plasticity, and the E/I balance. Across neurodevelopmental, neurodegenerative, psychiatric, and substance use disorders, altered PNN integrity and composition emerge as convergent mechanisms that link molecular dysfunction to behavioral pathology.
However, the nature and consequences of these alterations differ systematically across disease categories. In neurodevelopmental disorders such as ASD, the aberrant timing of PNN formation— whether premature, delayed, or excessively stabilized—disrupts critical period plasticity and inhibitory interneuron maturation, limiting experience-dependent circuit refinement. By contrast, in neurodegenerative disorders such as Alzheimer’s disease and ALS, PNN degradation is often secondary to neuroinflammatory processes, including microglial activation and extracellular matrix proteolysis, resulting in the destabilization of mature circuits and increased neuronal vulnerability. In disorders such as epilepsy, schizophrenia, and substance use disorders, PNN remodeling may initially reflect adaptive responses to altered activity but can ultimately promote maladaptive circuit rigidity or hyperexcitability. These comparisons highlight a unifying principle: PNN alterations are not inherently pathogenic or protective; rather, their impact depends on developmental timing, circuit context, and disease-specific perturbations.
Accordingly, therapeutic strategies targeting PNNs must be tailored to these contextual differences. Approaches such as enzymatic digestion, pharmacological inhibition of proteases, and genetic manipulation of ECM components, hold promise but require careful calibration (Figure 2). Challenges include achieving spatial precision, especially given regional and circuit-specific PNN functions, and defining optimal temporal windows for intervention. In neurodevelopmental disorders, therapies may aim to reopen or recalibrate rigid networks, whereas in neurodegeneration, preserving PNN-mediated stability may be paramount. In conditions with recurrent perturbations, such as epilepsy or addiction, timing relative to disease stage or relapse risk will determine therapeutic benefit.
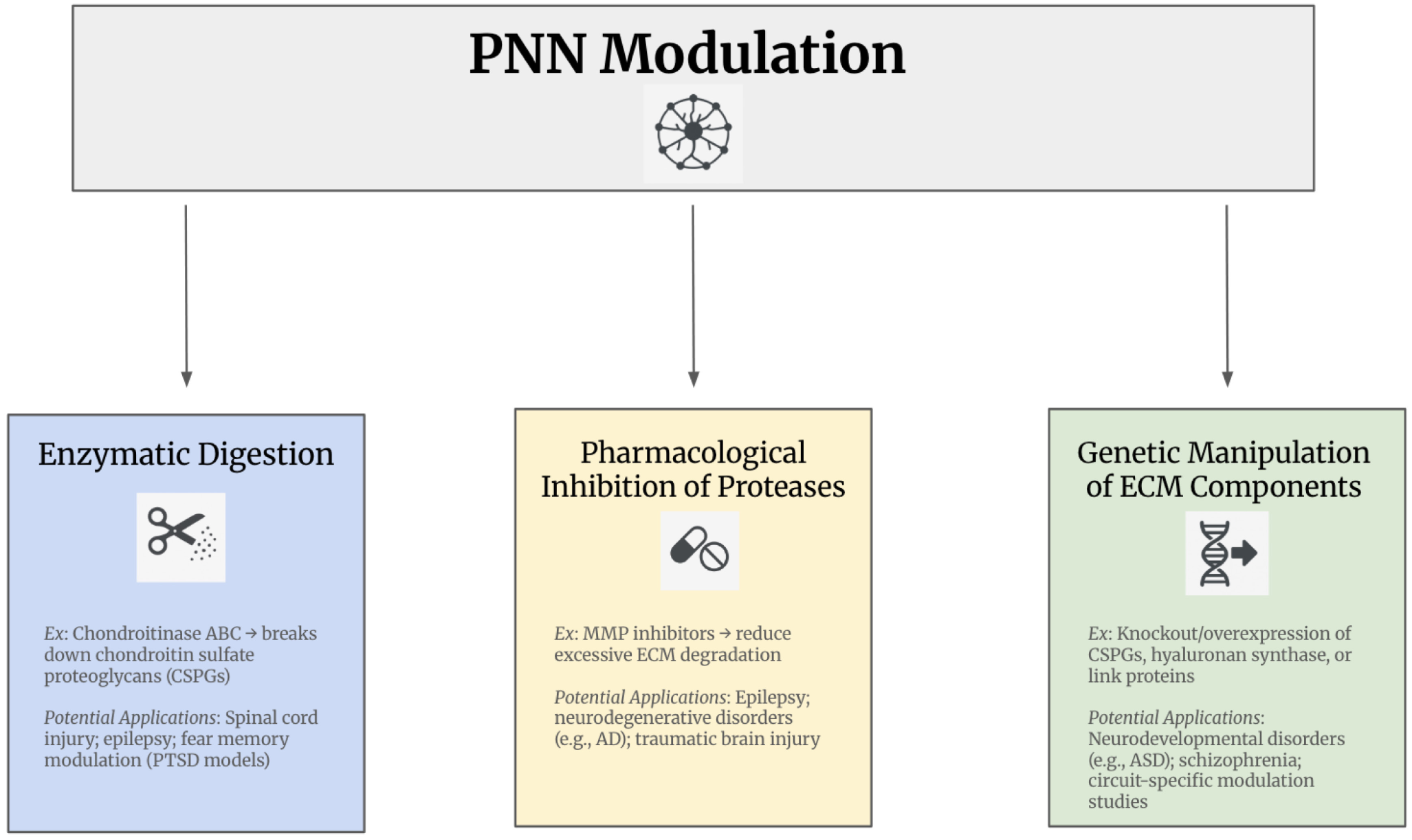
Therapeutic Targets of Perineuronal Net Modulation. Abbreviations: AD, Alzheimer’s disease; Autism spectrum disorder (ASD); CSPGs, Chondroitin sulfate proteoglycan; ECM, Extracellular matrix; MMP, Matrix metalloproteinases; PNN, Perineuronal net; PTSD, Post-traumatic stress disorder.
PNNs exemplify a paradoxical principle: they stabilize circuits and support function when appropriately localized and timed but can entrench maladaptive states when mistimed or excessive. Translational success will depend on integrating spatial precision, temporal specificity, and disease-context awareness. Future priorities should shift from descriptive mapping to strategy- driven approaches, including in vivo imaging and proteomics to track PNN dynamics, the identification of molecular regulators of PNN assembly and degradation, and the development of targeted therapies that modulate PNNs directly or via neuroinflammatory and activity-dependent pathways. Together, these considerations underscore the idea that PNN-targeted therapies cannot adopt a one-size-fits-all approach. Instead, successful translation will depend on integrating spatial precision, temporal specificity, and disease-context awareness into therapeutic design. Addressing these challenges will be essential for transforming PNN modulation from a powerful experimental tool into a viable clinical strategy.
Additionally, no studies have yet examined PNN alterations in individuals with neurological symptoms of long COVID. Given that immune dysregulation and neuroinflammation may contribute to cognitive impairment, assessing ECM/PNN dynamics in this context represents a critical avenue for research.
Overall, PNNs offer not only therapeutic potential but also promise as biomarkers for diagnosis, prognosis, and treatment response.
Their study represents a rapidly expanding frontier in neuroscience and psychiatry, with the potential to reshape our understanding of brain plasticity and disease.
Footnotes
Acknowledgements
The authors acknowledge the use of ChatGPT (model 3.5) developed by OpenAI for image creation of Figures 1 and ![]() . The author has reviewed and edited the content assisted by AI tools and assumes full responsibility for the content of the publication.
. The author has reviewed and edited the content assisted by AI tools and assumes full responsibility for the content of the publication.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author contribution
S.S. and O.G. performed the literature search, conceptualization, writing and validation.
Declaration of Conflicting Interests
The author declares no Conflicts of Interest.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Ethics Statement
Not applicable.
Informed Consent
Not applicable.