
Abstract
It has been reported that 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase inhibitors (statins) produce a variety of cardiovascular protective effects independent of their ability to lower total and low-density lipoprotein cholesterol. Recent studies have also reported that statins produce pleiotropic effects through improved endothelial function, enhanced fibrinolysis, and antithrombotic actions. In the present study, we examined the effects of pitavastatin, pravastatin, atorvastatin, and cerivastatin on endothelin (ET)-1 production in cultured porcine aortic endothelial cells (PAECs). Treatment with cerivastatin but not pitavastatin, pravastatin, or atorvastatin decreased basal and TNF-α–stimulated ET-1 release from PAECs in a dose-dependent manner (1–10 μM). Northern blot analysis showed that cerivastatin markedly suppressed prepro ET-1 mRNA expression in both conditions. In addition, these inhibitory effects of cerivastatin on ET-1 release and prepro ET-1 mRNA expression were completely abolished by simultaneous treatment with 200 μM mevalonate. Furthermore, cerivastatin did not have any effects on endothelial nitric oxide synthase (eNOS) protein levels, but induced eNOS phosphorylation at Ser1177. From these findings, it is most likely that cerivastatin suppresses ET-1 production, possibly through an increase in eNOS activity and the subsequent nitric oxide production in PAECs. These findings also suggest that cerivastatin may have beneficial effects on ET-1–related diseases.
Introduction
Endothelin (ET)-1 is a potent vasoconstrictor peptide purified from the supernatant of cultured porcine aortic endothelial cells (PAECs; Ref. 1) and possesses a number of biologic activities leading to vascular disorders (2). ET-1 biosynthesis and release seem to be regulated at the transcriptional level, because ET-1 release from endothelial cells (ECs) is constitutive. Several studies have indicated that various substances, such as thrombin (3), transforming growth factor-β1 (4), and tumor necrosis factor (TNF)-α (5), stimulate ET-1 gene expression in ECs by DNA binding of transcription factors, such as activator protein (AP)-1 and nuclear factor-1.
It has been demonstrated that a balance between ET-1 and nitric oxide (NO) production in
ECs plays a central role in maintenance of the integrity of vascular tone. NO, known as an
endothelium-derived relaxing factor, is formed from the terminal guanidino nitrogen atom of
3-Hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase inhibitors (statins) have been widely used for the treatment of hypercholesterolemia (10–13). Several clinical trials have demonstrated that statins are effective for preventing cardiovascular events, such as myocardial infarction, stroke, and sudden death (10–13). Accumulating evidence suggests that statins produce pleiotropic effects through improved endothelial function, enhanced fibrinolysis, and antithrombotic actions independent of their ability to lower total and low-density lipoprotein cholesterol (10, 11). In addition, it has been shown that various beneficial effects of statins may be caused by the activation of endothelial nitric oxide synthase (eNOS) and the subsequent increases in NO production in the vascular endothelium.
In the present study, we examined whether several statins (pitavastatin, pravastatin, atorvastatin, and cerivastatin) have an inhibitory effect on ET-1 production in cultured PAECs. Here, we show that cerivastatin but not pitavastatin, pravastatin, or atorvastatin markedly suppresses ET-1 production through the stimulation of eNOS phosphorylation.
Materials and Methods
Cell Culture.
All chemicals and reagents for cell culture were obtained from Invitrogen Corp. (Carlsbad, CA) except for fetal bovine serum (Biological Industries, Kibbutz Beit Haemek, Israel). PAECs were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere of 95% air and 5% CO2, as described (9). For all experiments, PAECs were grown to confluence and then made quiescent by incubation with serum-free DMEM containing 0.01% heat-inactivated bovine serum albumin for 12 hrs.
Radioimmunoassay for Determination of ET-1.
The radioimmunoassay for ET-1 was performed as described (9). ET-1 antiserum was kindly provided by Dr. M. R. Brown, University of California, San Diego and did not cross-react with big ET-1 (14).
Northern Blot Analysis.
Total RNA was isolated using the acid guanidium thiocyanate-phenol-chloroform extraction method. The isolated total RNA (5 μg per lane) was subjected to electrophoresis on a 1.1% agarose gel containing formaldehyde, and transferred to a nylon membrane. This membrane was hybridized with porcine prepro ET-1 cDNA probe (a gift from Dr. K. Goto, University of Tsukuba, Tsukuba, Ibaraki, Japan) and a GAPDH cDNA probe (BD Biosciences Clontech, Palo Alto, CA). Autoradiography was performed by exposing the membrane to imaging plates (Fuji Film, Tokyo, Japan). The autoradiograms of ET-1 were quantified by densitometric analyses, and signals of ET-1 mRNA were normalized for each sample, with respect to the density of the corresponding signal for GAPDH mRNA.
Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assay.
Nuclear extracts were prepared from PAECs using the method of Schreiber et al. (15). The nuclear extracts (2 μg protein) were used for the electrophoretic mobility shift assay, as described previously (9).
Western Blot Analysis.
All procedures were performed according to the method described elsewhere (9). The following commercially available antibodies were used: mouse antibody for eNOS (BD Pharmingen, San Diego, CA), rabbit polyclonal antibody for phospho-eNOS (Ser1177; Cell Signaling Technology, Beverly, MA), horse anti-mouse horseradish peroxidase–linked IgG for eNOS (Vector Laboratories, Burlingame, CA), and goat anti-rabbit horseradish peroxidase–linked IgG for phospho-eNOS (Zymed Laboratories, South San Francisco, CA).
Mevalonate.
Mevalonic acid lactone (Sigma-Aldrich Inc., St. Louis, MO) was modified to open the lactone ring by boiling at 50°C in a 0.1 mM NaOH solution for 1 hr. Afterward, the pH was adjusted to 7.4 with 0.1 mM HCl and aliquots were frozen at −80°C.
Statistical Analysis.
All values are expressed as mean ± SEM. For statistical analysis, we used one-way analysis of variance followed by Bonferroni’s multiple comparison tests. Differences were considered statistically significant at P < 0.05.
Results
Effect of Cerivastatin on ET-1 Production in PAECs.
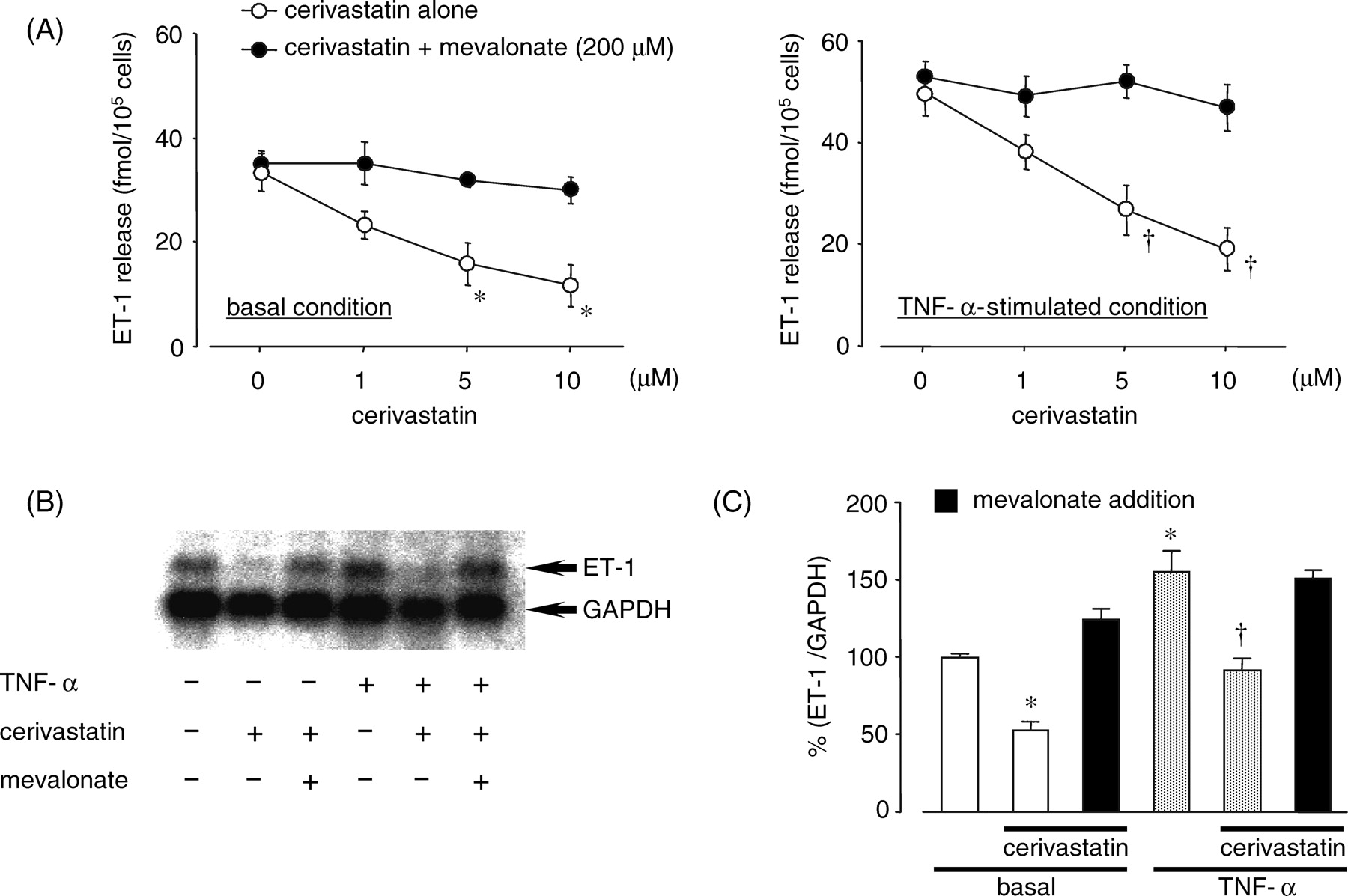
We first examined the effect of statins on ET-1 production in PAECs. Treatment with cerivastatin, but not pitavastatin, pravastatin, or atorvastatin, decreased basal and TNF-α–stimulated ET-1 release from PAECs in a dose-dependent manner (1–10 μM; Fig. 1). As shown in Figure 2, 10 μM cerivastatin decreased basal and TNF-α–stimulated prepro ET-1 mRNA expression. However, the inhibitory effects of cerivastatin on ET-1 release and prepro ET-1 mRNA expression in PAECs were completely abolished by simultaneous treatment with 200 μM mevalonate in basal and TNF-α–stimulated conditions.
Involvement of eNOS in the Regulation of ET-1 Production by Cerivastatin.
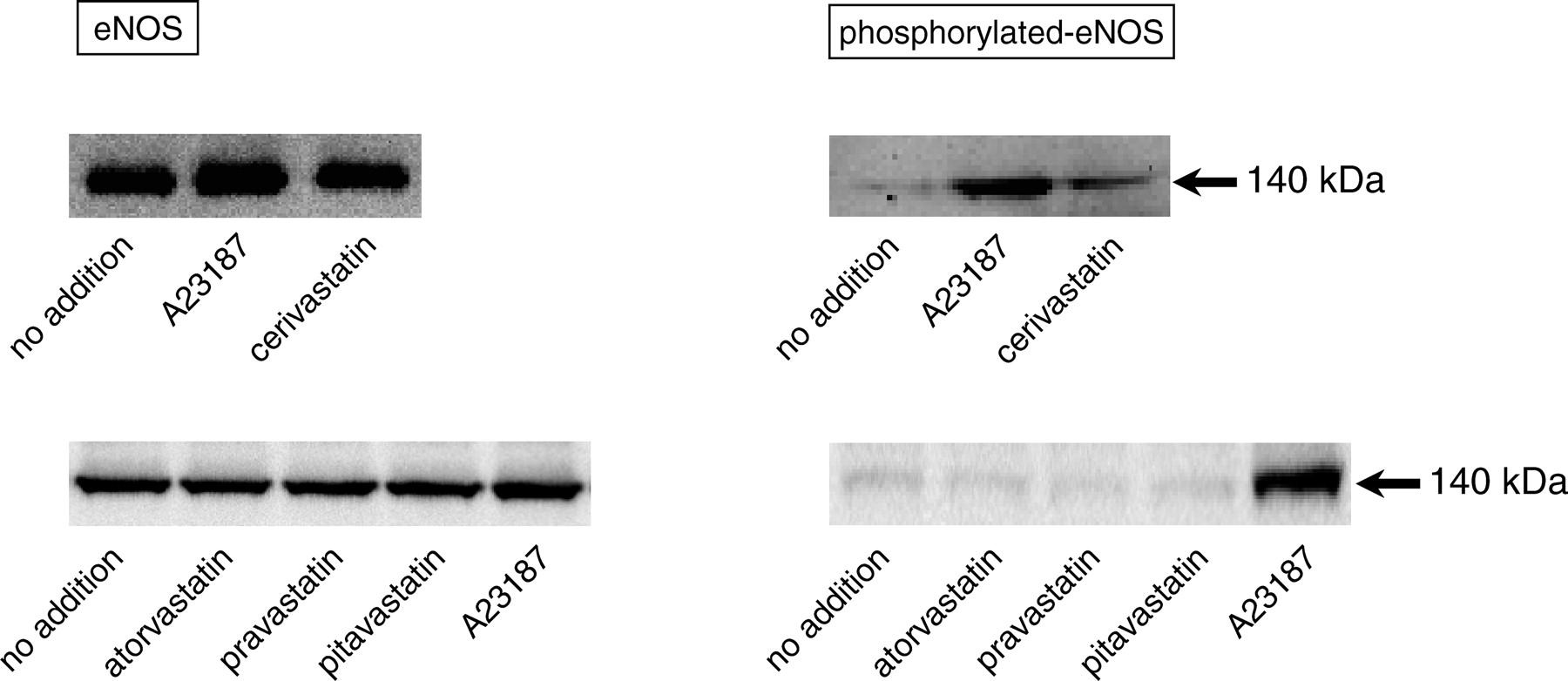
It is well known that various statins induce NO production through the upregulation of eNOS expression in ECs (11). Our previous studies have demonstrated that endogenous NO regulates ET-1 production at a transcriptional level in ECs (8, 9). Thus, we investigated whether cerivastatin affects ET-1 production through the regulation of eNOS and/or phosphorylated eNOS protein expression in PAECs. For detection of phosphorylated-eNOS protein in this experiment, we used a calcium ionophore, 10 μM A23187, as a positive control. As shown in Figure 3, addition of A23187 significantly increased both eNOS and phosphorylated eNOS protein levels in PAECs. Although no statins had an effect on the eNOS protein levels, only cerivastatin augmented phosphorylated eNOS protein levels. However, it has been reported that the phosphatidylinositol 3-kinase (PI3-kinase) pathway, which activates the serine/threonine protein kinase, Akt, enhances eNOS phosphorylation (16–18). Thus, we examined the effect of the PI3-kinase inhibitor, LY294002, on ET-1 release from PAECs under cerivastatin treatment. LY294002 dose-dependently suppressed the cerivastatin-induced decreases in ET-1 release from PAECs (data not shown).
Discussion
In the present study, we showed that cerivastatin suppressed ET-1 release from PAECs in both basal and TNF-α–stimulated conditions. Simultaneously, we found that these effects were accompanied by reduced prepro ET-1 mRNA expression. In addition, these inhibitory effects of cerivastatin on ET-1 production were completely abolished by combination with mevalonate. Thus, our results clearly suggest that cerivastatin inhibits ET-1 production in ECs through the suppression of HMG-CoA reductase.
There are some reports indicating that several statins, such as simvastatin, atorvastatin, and pitavastatin, suppress ET-1 production in cultured ECs (19, 20). The inhibitory effects of these statins were accompanied by the increase in eNOS mRNA and protein levels. Although we also examined the effects of pravastatin, atorvastatin, and pitavastatin on ET-1 production, these statins failed to attenuate the ET-1 release from PAECs or enhance the eNOS protein levels in PAECs. The reason for these different efficacies between cerivastatin and others remains unclear, but it may be because of the differences in the membrane permeability of each statin and the experimental methods, including cell type and species used.
Recent studies have demonstrated that many stimuli (including insulin, vascular endothelial growth factor, and β-agonists) induce NO production by activating eNOS via Ser1177 phosphorylation through the PI3-kinase/Akt pathway (21–23), suggesting that the phosphorylation status of eNOS at Ser1177 has important implications in its enzymatic activity. In the present study, we observed that treatment of cerivastatin, but not pitavastatin, pravastatin, or atorvastatin, enhanced eNOS phosphorylation in PAECs. We also noted that treatment with a PI3-kinase inhibitor, LY294002, abolished the decrease in ET-1 release from PAECs treated with cerivastatin. Taken together, it is most likely that cerivastatin-induced enhancement of eNOS phosphorylation through the PI3-kinase/Akt pathway and the subsequent increase in NO production may result in the suppression of ET-1 production in PAECs. Furthermore, the reason why other statins, except cerivastatin, failed to reduce ET-1 production in our experimental conditions may be attributable to the different effect on eNOS phosphorylation in PAECs.
Several studies have indicated that the ET-1 gene is regulated by a variety of transcriptional factors, such as AP-1 and GATA-2 (2). We and others have recently demonstrated that a transcriptional factor, nuclear factor-κB (NF-κB), is also responsible for the regulation of ET-1 gene expression (24–26). This view is based on findings that various NF-κB suppressors can decrease ET-1 production in cultured ECs. Moreover, a binding sequence for the activated NF-κB is located in the promoter region of the ET-1 gene. However, we recently found that NO can suppress ET-1 production through the regulation of NF-κB activation (9). We speculated that cerivastatin-induced NO production may affect NF-κB activation and the subsequent ET-1 gene expression. When we examined the effects of cerivastatin on NF-κB activation pathway, cerivastatin had no inhibitory effects on basal and TNF-α–induced NF-κB activation, inhibitor κB-α (IκBα) phosphorylation, and IκBα degradation in PAECs (data not shown). Thus, these observations suggest that the reduction of ET-1 production by cerivastatin is independent of the NF-κB activation pathway.
In conclusion, we demonstrated that cerivastatin suppressed ET-1 production in cultured PAECs, possibly through the activation of eNOS phosphorylation. These findings suggest that cerivastatin may have beneficial effects on ET-1–related diseases.
Effects of statins on ET-1 release from cultured PAECs. PAECs were pretreated with
the indicated concentrations of various HMG-CoA reductase inhibitors (cerivastatin,
pitavastatin, atorvastatin, and pravastatin) for 24 hrs, then washed and further
incubated with or without 10 ng/ml TNF-α for 6 hrs. The amount of ET-1 in the culture
medium was measured using a radioimmunoassay. Each point and bar represents the mean ±
SEM (n = 6). *P < 0.01;
†P < 0.01; compared with no addition. Effects of cerivastatin and mevalonate on basal and TNF-α–induced ET-1 release (A)
and prepro ET-1 mRNA expression (B and C). (A) PAECs were pretreated
with the indicated concentrations of cerivastatin in the absence or presence of 200
μM mevalonate for 24 hrs and then washed and further incubated with
or without 10 ng/ml TNF-α for 6 hrs. Each point and bar represents the mean ± SEM
(n = 6). *P < 0.01; †P <
0.01, compared with no addition, respectively. (B and C) PAECs were pretreated with 10
μM cerivastatin in the absence or presence of 200
μM mevalonate for 24 hrs, then washed and further incubated with or
without 10 ng/ml TNF-α for 2 hrs. Each column and bar represents the mean ± SEM
(n = 3). *P < 0.01, compared with no addition;
†P < 0.01, compared with TNF-α alone. Effects of statins on eNOS in cultured PAECs. PAECs were incubated with 10
μM of cerivastatin, atorvastatin, pravastatin, or pitavastatin for
24 hrs. The cell lysate was subjected to sodium dodecyl sulfate polyacrylamide gel
electrophoresis followed by Western blot analysis with specific antibodies against
eNOS (left panel) and phosphorylated-eNOS (at Ser1177; right panel). The cell lysate
from PAECs treated with a calcium ionophore, 10 μM A23187, was used
as a positive control for detection of eNOS and phosphorylated-eNOS protein.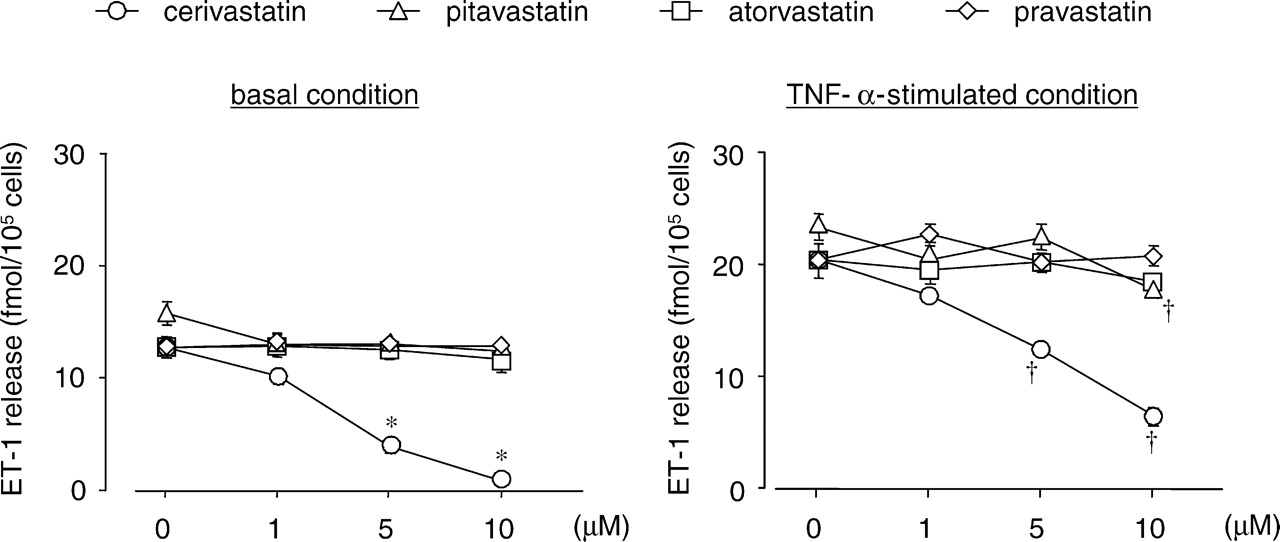
Footnotes
This work was supported by a “High-Tech Research Center” Project for Private Universities: matching fund subsidy from MEXT (Ministry of Education, Culture, Sports, Science and Technology), 2002–2006.