
Abstract
Regulation of ion channels by heterotrimeric guanosine triphosphatases (GTPases), activated by heptathelical membrane receptors, has been the focus of several recent reviews. In comparison, regulation of ion channels by small monomeric G proteins, activated by cytoplasmic guanine nucleotide exchange factors, has been less well reviewed. Small G proteins, molecular switches that control the activity of cellular and membrane proteins, regulate a wide variety of cell functions. Many upstream regulators and downstream effectors of small G proteins now have been isolated. Their modes of activation and action are understood. Recently, ion channels were recognized as physiologically important effectors of small GTPases. Recent advances in understanding how small G proteins regulate the intracellular trafficking and activity of ion channels are discussed here. We aim to provide critical insight into physiological control of ion channel function and the biological consequences of regulation of these important proteins by small, monomeric G proteins.
Introduction
Small guanosine triphosphatase (GTPase)-binding proteins are monomeric GTPases with molecular masses ranging from 20 kDa to 40 kDa. Typically, these proteins have much sequence homology and share several conserved domains, including consensus amino acid sequences responsible for interaction with guanosine diphosphatase (GDP) and GTP, and a region interacting with downstream effectors. Moreover, small G proteins belonging to the Ras, Rho, and Rab families have sequences at their carboxylic acid termini that undergo post-translational processing, including lipidation by addition of farnesyl, geranylgeranyl, palmitoyl, and proteolysis. More than 100 small G proteins have been identified in eukaryotes (1–3). The members of this superfamily are structurally classified into, at least, 5 families: (i) Ras, (ii) Rho, (iii) Rab, (iv) Sar1/Arf, and (v) Ran. Generic definitions for family functions have been developed; however, we must guard against restricting understanding of function to these narrow limits. Ras subfamily members primarily regulate gene expression. Rho subfamily members regulate both cytoskeleton reorganization and gene expression. Rab and Sar1/Arf family members regulate intracellular vesicle trafficking. Ran family members regulate nucleocytoplasmic transport during G1, S, and G2 phases of the cell cycle and microtubule organization during the M phase (3).
Small G proteins involved in signal transduction, similar to heterotrimeric G proteins, are present only in eukaryotes from yeast to human, although unrelated G proteins involved in protein synthesis (e.g., elongation factors) exist in both prokaryotes and eukaryotes. Most small G proteins are widely distributed in mammalian cells, with most cells having at least 1 representative each of the Ras, Rho, Rab, Sar1/Arf, and Ran families, although expression levels of their members vary. A few members, though, show tissue-specific expression. For example, Rab17 is detected only in epithelial cells (4, 5). Most small G proteins are localized either in the cytosol or at plasma and intracellular membranes. Usually, each type of small G protein is localized to a specific membrane (3). For instance, Ras proteins localize to the cytoplasmic face of the plasma membrane. This localization is mediated, in part, by post-translational lipidation and proteolysis. Rho proteins, in contrast, dynamically translocate between the plasma membrane and cytosol. Most Rab proteins through geranylgeranylation of Cys-X-Cys and Cys-Cys motifs localize to the cytoplasmic faces of plasma and intracellular membrane compartments (3). Recently, Heo et al., imaging the subcellular location of fluorophore-tagged small GTPases in cultured mammalian cells, surveyed plasma membrane–targeting mechanisms. Approximately half (48 of 125) of the small G proteins assayed were fully or partially localized to the plasma membrane (6).
Small G proteins act as GTP-dependent switches to control the activity of effector proteins and thus initiate cellular signaling cascades. These proteins reside in 2 states: the GDP-bound inactive and the GTP-bound active forms. An upstream signal stimulates the dissociation of GDP from the GDP-bound form, which is followed by the binding of GTP, leading to a conformational change favoring effector binding. Discrete small G proteins are often capable of directly interacting with several first effectors. GTP is metabolized by an inherent GTPase activity, which can be modified by accessory proteins and effectors. The interaction between small G protein and effector leads to changes in activity of the latter (3).
Evidence that ion channels are final effectors of small G proteins has accumulated for the last decade. Signaling mediated by small G proteins can impinge upon the activity of a wide variety of membrane-resident ion channels. In some cases, small GTPases interact directly with ion channels to elicit regulation, and in others, regulation is mediated by intermediary signaling proteins. For example, Rab11a directly binds the epithelial Ca2+ channel (7) and Rem forms a regulatory complex with the Ca2+ channel auxiliary β-subunit (CaVβ) (8). Similarly, RhoA physically associates with the amino terminus of Kv1.2 (9). In contrast, K-Ras and RhoA increase the activity of the epithelial Na+ channel (ENaC) via PI3-kinase and PI(4)P5-kinase signaling pathways (10, 11). Similarly, Rac1 mediates rapid insertion of transient receptor potential channel 5 (TRPC5) channels into the plasma membrane through stimulation of PI(4)P5-kinase (12).
Here we discuss recent results supporting functional interactions between ion channels and small G proteins, as well as detail the molecular mechanisms underpinning such interactions and discuss the biological consequences of this regulation. We concentrate on regulation of ion channels by small G proteins in the Ras, Rho, and Rab families (Tables 1–3 ), as well as document some of the best-studied examples of such regulation.
Regulation of Ion Channels by the Ras Family
There are 4 major Ras isoforms: Ha-Ras, Ki-RasA, Ki-RasB, and N-Ras. Ras proteins bind to and activate the serine/threonine kinase Raf (13), which then initiates the mitogen-activated protein kinase (MAPK) cascade to modulate gene expression. Various extracellular signaling molecules, notably growth factors and other agonists of receptor tyrosine kinases, initiate this signaling cascade (14–16). A variety of candidate Ras protein effectors, in addition to Raf, have been reported. There is good support for Ras proteins also binding and activating Ral GDS, RIN1, and PI3-kinase (17–19). Thus, Ras proteins are positioned to initiate a complex arrangement of cellular signaling cascades.
Ras activity is regulated by positive and negative regulators: GTPase-activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs), respectively. Phospho-tyrosines within receptor tyrosine kinases serve as docking sites for adaptor proteins, such as Grb2, which, in turn, recruit Ras GEFs, such as SOS, to produce a receptor-adaptor-GEF complex capable of attracting and activating Ras. Receptors not directly associated with tyrosine kinases may also activate Ras proteins indirectly through Src-like tyrosine kinases or ligand-independent activation of receptor tyrosine kinases (3). Moreover, heterotrimeric G protein-coupled receptors, such as muscarinic acetylcholine receptors, α-adrenergic receptors, and lysophosphatidic acid receptors, have also been shown to activate Ras proteins (20–23).
Signaling mediated by Ras and other Ras-like small G proteins can impinge upon the activity of membrane-resident ion channels. For instance, K-RasA increases the activity of ENaC (24, 25). We (14, 26) and others (27–29) demonstrated that in renal epithelia, aldosterone via control of gene expression increases the levels and activity of the small G protein K-Ras. K-Ras increases the activity of channels present in the membrane via PI-3 kinase signaling by increasing channel open probability (24, 25, 27). Regulation of ENaC open probability by PI3-kinase is a direct consequence of the physical association of the product of PI3-kinase, PI(3,4,5)P3 with the channel. Overexpression of K-Ras with ENaC increases channel activity independently of effects on ENaC membrane levels. K-Ras actions on ENaC are blocked by the PI3-kinase inhibitor wortmannin and mimicked by overexpression of active PI3-kinase. Overexpression of an effector-specific mutant of Ras capable of only activating PI3-kinase increases ENaC activity in a wortmannin-sensitive manner. Together, these observations reveal that K-Ras activates ENaC via PI3-kinase signaling with concomitant increases in PI(3,4,5)P3 levels. ENaC activity is limiting for Na+ absorption across many epithelia, including that responsible for negative feedback control of blood volume and pressure (30–33). Thus, Ras regulation of ENaC may play a role in control of systemic Na+ balance and blood pressure.
Distinct members of the Ras superfamily can, in some instances, have isoform-specific effects on ion channel activity, as well as have different effects on distinct channels. For example, Ha-Ras via MAPK signaling decreases the activity of inward rectifier K+ channel 1 (IRK1) by promoting retrieval of the channel from the plasma membrane (34) but increases the activity of T-type Ca2+ channels (35). Small G proteins within the RGK (Rad, Rem, Rem2, Gem/Kir, Ras-related protein) subfamily directly interact with the β-subunit of L-type Ca2+ channels, promoting retrieval from the plasma membrane and decreased activity (8, 36–38). However, Rem2 reduces N-type Ca2+ channel activity without affecting channel surface density (39). The intracellular trafficking of Ca2+ channels from their site of synthesis in the endoplasmic reticulum to the cell surface is an absolute requirement for their function, regardless of their specific site of action. Many ion channel subunits contain unique trafficking signals that post-translationally control expression. Beguin et al. (36) took the first step in investigating how Ca2+ channel β subunits might be dynamically modulated through protein-protein interactions. In the process, they uncovered a link between regulation of Ca2+ channel trafficking (and therefore plasma membrane electrical properties) and signaling by small G proteins. Using the Ca2+ channel auxiliary β3 subunit as bait to screen a cDNA library, they identified the Gem/Kir as an interacting partner. Remarkably, the binding of Kir/Gem to ß subunits inhibited their assembly with α1 subunits, such that Gem/Kir coexpression eliminates Ca2+ channel expression and, importantly, the Ca2+-dependent process of secretion.
Rap and Ras also have opposing actions on atrial K+ channels. Activation of M2-muscarinic receptor-coupled K+ channels in the heart is inhibited by the addition of exogenous Ras and Ras-GAP (40). Interestingly, Rap1A, which is a Ras-related GTP-binding protein, antagonizes the ability of Ras and Ras-GAP to inhibit muscarinic K+ channels (41). Similarly, Ras and Rap1 function as opposing regulators of voltage-gated sodium currents (42), N-methyl-
Thus, several ion channels, such as ENaC, IRK1, and L-, N-, and T-type Ca2+ channels, are final effectors of signaling cascades initiated by Ras proteins. We can conclude that Ras small G proteins are capable of regulating the activity of many types of ion channels, albeit often through distinct signaling cascades.
Regulation of Ion Channels by Rho Family Small G Proteins
Rho GTPases constitute a distinct family within the superfamily of Ras-related small GTPases and are found in all eukaryotic cells. The most extensively characterized members are Rho, Rac, and Cdc42. Small G proteins in the Rho family are known to regulate diverse cellular processes, including cytoskeletal organization, cell proliferation, gene transcription, and cell cycling. Given the involvement of Rho small G proteins in such a wide variety of important cellular processes, it is not surprising that a great deal of effort has been put into identifying their cellular targets or effector proteins. To date, 30 or so potential effectors of Rho, Rac, and Cdc42 have been identified (45). Recently, Rho GTPase signaling also has been shown to modulate the activity of several classes of ion channels, including Kv1.2 and ERG K+ channels, Ca2+ channels, and epithelial Na+ channels, as well as nonselective cation channels (Table 2) (9, 10, 46–50). Unfortunately, little is currently known of the upstream regulators of Rho small GTPases (3, 51).
Similar to other small G proteins, Rho GTPases act as molecular switches cycling between active GTP-bound and inactive GDP-bound states. In the GTP-bound state, Rho GTPases interact with effector molecules, such as Rho kinase, WASP, PI(4)P5-kinase, and PI3-kinase, to initiate downstream responses. Both RhoA and Rac1 as well as Rho-kinase increase PI(4,5)P2 levels by activating PI(4)P5-kinase (52–55). Rac and Cdc42 also interact with PI3-kinase and increase PI(3,4,5)P3 levels (56–58). Through these phospholipid second messengers and adapter proteins, Rho small G proteins influence vesicle movement affecting both endocytosis and exocytosis (59–62).
The effects of RhoA on ENaC are similar to those described for K-Ras in the sense that they both increase channel activity. However, their transduction pathways and mechanism activation are different (11); K-Ras increases channel open probability, whereas RhoA increases channel membrane levels. RhoA activates ENaC via Rho-kinase and subsequent activation of PI(4)P5-kinase with concomitant increases in PI(4,5)P2 levels promoting channel insertion into the plasma membrane (10, 47). It is interesting that phospholipids appear to serve as signaling intermediates in both instances. Shown in Figure 1 are a summary graph and possible models for regulation of ENaC by K-Ras and RhoA. Similar to RhoA actions on ENaC, Rac1, another small G protein in the Rho family, promotes insertion of TRPC5 into the plasma membrane (12). This effect on TRPC5 is dependent on Rho-kinase signaling and PI(4)P5-kinase activity, again implicating PI(4,5)P2 as playing a critical role. Rac1 was shown to be both necessary and sufficient to stimulate the rapid incorporation of functional TRPC5 channels into the plasma membrane through activation of PI(4)P5-kinase. The current findings show a similar transduction pathway and mechanism of action of small G proteins in the Rho family on ENaC and TRPC5.
Proteins in the Rho family are involved in the regulation of F-actin polymerization. Rho inhibits cAMP-dependent translocation of the water channel aquaporin-2 (AQP2) into the apical membrane of epithelial cells by controlling the organization of the actin cytoskeleton (62, 63). RhoA promotes ENaC trafficking to the plasma membrane likely through effects on the microtubules but not on the microfilament cytoskeleton (64). Rho signaling and its effects on epithelial ion channels to include control of ENaC and AQP2 membrane levels may be part of a larger cellular program tightly controlling epithelial transport.
The voltage-activated potassium channel ether-a-go-go–related gene channel (KCNH2 or ERG) is essential for normal electrical activity in the heart (65, 66). Rac1 and RhoA mediate opposing hormonal regulation of ERG channels. Rho inhibits this channel via a protein serine/ threonine (S/T) kinase, whereas Rac stimulates the channel via a protein S/T phosphatase (49). Recently, it was shown that, the S/T phosphatase PP5 acts as a direct molecular effector for activated Rac (46). Although Rho and Rac signaling pathways interact at many levels, Rac-dependent stimulation of PP5 provides a direct molecular mechanism for the antagonism of Rho-dependent signaling.
Rho and Rho-kinase are also involved in lysophospha-tidylcholine (LPC)-induced nonselective cation current (48). LPC is an amphipathic metabolite of membrane phosphatidylcholine; high concentrations of LPC have been found in ischemic hearts. Therefore, LPC is thought to be one of the causes of Ca2+ overload and arrhythmia during cardiac ischemia and reperfusion. Thus, regulation of nonselective cation currents by small G proteins may contribute to this pathology.
Together, the results described above show that many small G proteins in the Rho family are involved in ion channel modulation. However, although the role of Rho GTPases in regulation of ion channels has been recognized, the underlying common mechanisms are far from understood. The regulation of ion channels by Rho proteins is immensely complex and is modulated by many factors, including cross-talk with other intracellular signaling pathways, interactions with cytoskeletal elements, and probably other factors as yet uncharacterized. As investigations into this remarkable field continue, it is likely that further details and additional regulatory mechanisms will be uncovered.
Regulation of Ion Channels by Rab Family Small G Proteins
Rab proteins exist in all eukaryotic cells and constitute the largest branch of the small G protein superfamily. In mammalian cells, more than 60 Rab proteins have been identified (3, 67, 68). A large body of evidence has accumulated in support of a role for Rab proteins in vesicle trafficking from yeast to human. Rab proteins use the guanine nucleotide-dependent switch mechanism to regulate each of the 4 major steps in intracellular vesicle transport: (i) vesicle budding from the donor membrane, (ii) targeting of the vesicle to the acceptor membrane, (iii) docking of the vesicle, and (iv) fusion of the vesicle with the acceptor membrane. Substantial evidence supports that most Rab proteins regulate the targeting/docking/fusion processes and that only some of them regulate the budding process, which is primarily regulated by Sar1/Arf small G proteins (3). Recently, several groups have demonstrated that Rab proteins play a key role in the modulation of various ion channels. Although still far from being completely understood, these studies provide crucial insights into how Rab proteins might be involved in the distribution of ion channel proteins between different compartments in the cell and the plasma membrane (69).
Van de Graaf et al. (7) identified Rab11a as a novel TRPV5- and TRPV6-associated protein. Rab11a colocalizes with TRPV5 and TRPV6 in Ca2+-transporting epithelial cells of the kidney. Here, both Rab11a and TRPV5 are present in vesicular structures underlying the apical plasma membrane (70). TRPV5 and TRPV6 preferentially interact with Rab11a in its GDP-bound conformation. Co-expression of a mutant Rab11a protein, locked in the GDP-bound state, resulted in significantly decreased Ca2+ uptake caused by diminished channel cell surface expression, indicating a direct role of Rab11a in the trafficking of TRPV5/6 toward the plasma membrane. A helical stretch in the carboxyl terminus containing 5 amino acids at position 595–601 within the Rab11a-binding site of TRPV5 was demonstrated to be required for Rab11a binding. Moreover, this region is conserved among all identified species of TRPV5 and TRPV6. Other Rab GTPases, including Rab7 and Rab22b, did not bind TRPV5 and TRPV6, indicating the specificity of the interaction of TRPV5/6 with Rab11a (7). Therefore, it is likely that epithelial Ca2+ channels TRPV5 and TRPV6, present on the plasma membrane, come from intracellular endosomes in a Rab11-dependent manner.
Rab 3, Rab 4, and Rab27a inhibit ENaC activity via protein-protein interactions (71, 72). Rab27a inhibits ENaC function through a complex mechanism involving Munc13–4 (a putative priming factor for exocytosis) and SLP-5 (synaptotagmin-like protein) (73). Similarly, Rab27a and Rab4 negatively regulate CFTR function (74, 75). Rab5 regulates the trafficking of CFTR from the plasma membrane to early endosomes. From early endosomes, CFTR is recycled back to the plasma membrane through recycling endosomes that are controlled by Rab11 and Rme1 (76). Rab7 controls the movement of CFTR from early endosomes to late endosomes and also facilitates the trafficking of CFTR to lysosomes. Rab9, in comparison, can drive CFTR from late endosomes to the trans-Golgi network. From the trans-Golgi, CFTR can reenter the secretory pathway and be delivered to the plasma membrane. Thus, Rab proteins play a role in CFTR recycling to the plasma membrane through recycling endosomes and via the trans-Golgi network. Regulation of CFTR trafficking by Rab has been exploited to increase the plasma membrane expression level of ΔF508-CFTR, the most common mutation in CFTR (77, 78) causing cystic fibrosis due to diminished membrane levels of this channel. Either inhibition of Rab5-dependent internalization or augmentation of a Rab11-dependent recycling step results in increased plasma membrane pools of ΔF508-CFTR, demonstrating the physiological importance of regulation by small G proteins (77).
Conclusions
The study of ion channels and small G proteins has expanded considerably in recent years. It is now clear that many ion channels are targets of small G proteins and their associated downstream signaling cascades. Small G proteins modulate ion channel activity by influencing channel open probability and/or channel trafficking to the plasma membrane.
Although all small G proteins serve as molecular switches, their final effects on ion channels are variable, with some being positive regulators and others being negative regulators. Moreover, distinct small G proteins often have inverse effects on a particular ion channel. In addition, regulation of ion channels by small G proteins appears to be widespread because several specific small G proteins are capable, albeit often through distinct signaling mechanisms, of modulating diverse channel types. This large array of small G protein regulators and channel effectors enables a cell to respond in a physiologically appropriate manner to locale and systemic signaling inputs. Undoubtedly, many questions remain regarding regulation of ion channels by small G proteins.
Regulation of Ion Channels by Small Guanosine Triphosphatases (GTPases) in the Ras Family
Regulation of Ion Channels by Small Guanosine Triphosphatases (GTPases) in the Rho Family
Regulation of Ion Channels by Small Guanosine Triphosphatases (GTPases) of Rab Family
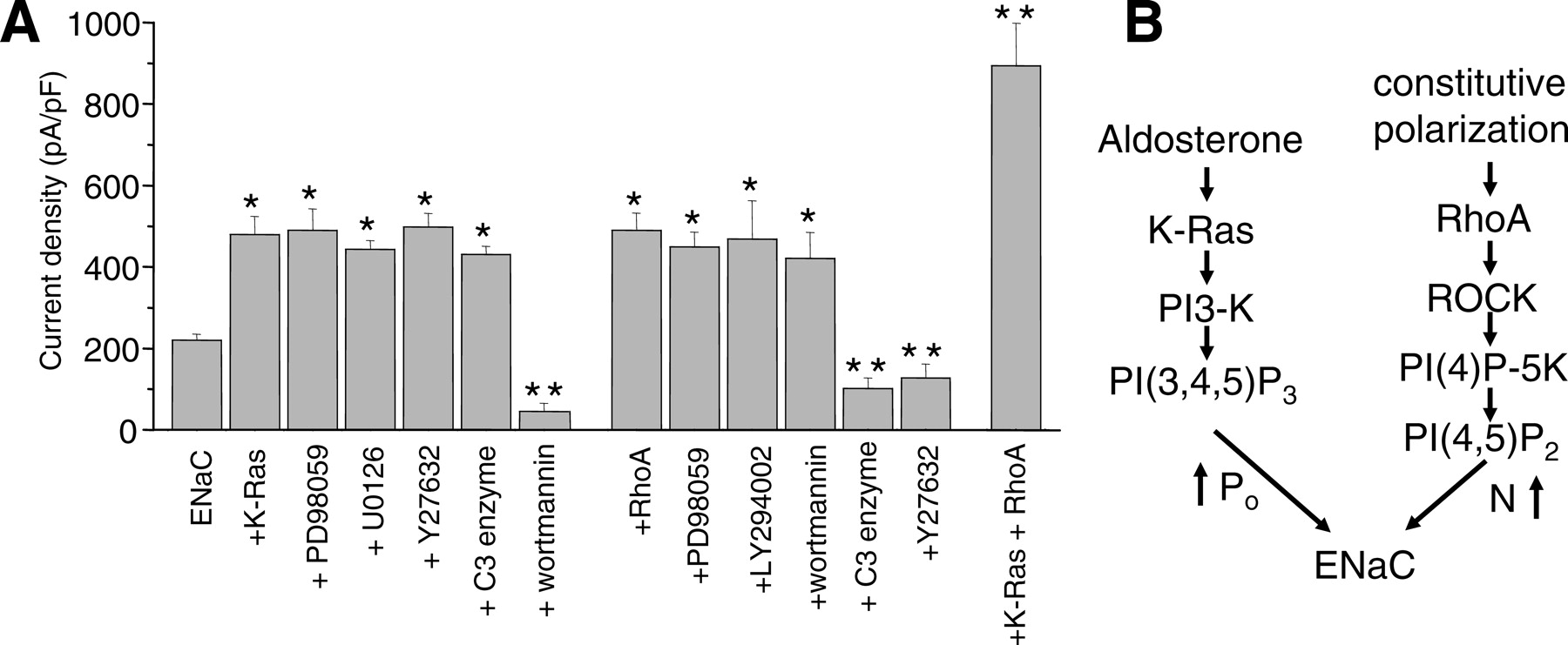
K-Ras and RhoA have independent actions on epithelial Na+ channel. (A) This summary graph shows the relative change in activity of ENaC plus K-Ras or RhoA alone or together without and with pretreatment with the PI3-K (wortmannin and LY294002), MAPK (PD98059 and U0126), or Ral/Rac/Rho (Rho-kinase inhibitor Y27632; Rho inhibitor C3 exoenzyme included in pipette solution) signaling inhibitors. Data presented as mean ± SEM amiloride-sensitive current density at −80 mV for voltage-clamped Chinese hamster ovary cells expressing mENaC in the absence and presence of small G proteins. *, P < 0.05 versus mENaC alone; **, P < 0.05 versus mENaC plus K-Ras and/or RhoA. (B) Models illustrating possible mechanisms by which K-Ras and RhoA increase ENaC activity. Some results, presented here in a different format, were published previously in (10, 11, 24, 47).
Footnotes
Acknowledgements
Research by J.D. Stockand and A. Staruschenko was supported by the National Institutes of Health, the American Heart Association, and the National Kidney Foundation.