
Abstract
Although Orai1 protein was recently identified as the component of CRAC channels in hematopoietic cells, store-operated channels (SOC) in other cell types may have a different molecular entity. Also, the activation mechanism of SOC remains unclear, in general. In the present study, we tested the hypothesis that TRPC1 and TRPC4 proteins were functional subunits of SOC in glomerular mesangial cells (MCs) and that STIM1 was required for the channel activation through interaction with the TRPC proteins. In cultured human MCs, cell-attached patch clamp and fura-2 fluorescence measurements showed that single knockdown of either TRPC1 or TRPC4 significantly attenuated thapsigargin-induced membrane currents and Ca2+ entry as well as Ang II–induced channel activity. Double knockdown of both TRPCs resulted in a comparable inhibition on store-operated Ca2+ entry with single knockdown of either TRPC. Consistent with our previous report, co-immunoprecipitation showed a physical interaction between TRPC1 and TRPC4. Furthermore, we found that knockdown of STIM1 using RNAi significantly reduced the thapsigargin-stimulated membrane currents. Co-immunoprecipitation showed that STIM1 interacted with TRPC4, but not TRPC1. In addition, simultaneous inhibition of STIM1 and TRPC1 resulted in a comparable effect on SOC with single inhibition of either one of them. Taken together, we conclude that in glomerular mesangial cells, the TRPC1/TRPC4 complexes constitute the functional subunits of SOC and that the interaction between STIM1 and TRPC4 may be the mechanism for the activation of the channels.
Introduction
Glomerular mesangial cells (MCs) are located within glomerular capillary loops and contribute to the physiological regulation of glomerular hemodynamics (1, 2). Ca2+ influx via plasma membrane is a major component of the response of MCs to hormones (1–4). We and others have recently demonstrated that store-operated Ca2+ channel (SOC) is an important mechanism for the Ca2+ response induced by vasoactive agonists, such as Ang II or endothelin, in cultured MCs (3–6).
Store-operated Ca2+ entry (SOCE) is a ubiquitous signaling mechanism in both non-excitable and excitable cells and is responsible for the regulation of diverse cellular functions ranging from cell proliferation and gene expression to cell contraction and secretion (7). Physiologically, this Ca2+ entry mechanism is triggered by activation of phospholipase C by G-protein–coupled receptors or receptor tyrosine kinases that induces Ca2+ release from the endoplasmic reticulum (ER), which then activates store-operated Ca2+ channels (SOCs) in the plasma membrane. Despite intensive studies in the last 2 decades, the molecular identity of the SOC and the cellular mechanisms underlying the coupling of store depletion and Ca2+ entry remain elusive. This case is even more complicated by the fact that SOC behaves differently in different types of cells, implying that the protein components or regulatory mechanisms of SOC might be cell-type specific. For instance, the SOC of lymphocytes, known as Ca2+ release-activated Ca2+ channels (CRAC), are distinguished from other SOCs by their extremely high selectivity for Ca2+ and extremely low conductance (8, 9). However, these properties are not displayed in excitable cells (10–12).
Members of the canonical transient receptor potential (TRPC) protein family have been proposed as candidates for SOC (7, 13). These include studies on TRPC1 (14, 15), TRPC3 (16), TRPC4 (17), TRPC5 (18), TRPC6 (19), and TRPC7 (20). TRPCs are thought to exist as tetramers and, in many cases, different members within TRPC subgroups may exist as functional heteromultimers (13, 21, 22). For example, SOCE is mediated by TRPC1 complex forming with TRPC3 in human parotid gland ductal cells and hippocampal neuronal cells, or with TRPC4 in endothelial cells (23–25). This variability of subunit complex composition in different tissues may confer specificity of channel function. Recently, a major breakthrough in the field of SOC is the finding that Orai1 protein is the component, most likely the pore-forming subunit of CRAC channel, the prototypic SOC (26–31). However, whether the Orai genes account for all SOCs existing in different cell types and whether the Orai1 protein requires binding partners to form native SOCs remain unknown.
Recently, the stromal interaction molecule 1 (STIM1) has been identified as a Ca2+ sensor in the ER, activating SOC at the plasma membrane upon store-depletion (32, 33). There is evidence that STIM1 diffusely distributed in the ER membrane translocates to the regions of the ER directly apposed to the plasma membrane upon store depletion, where it triggers SOCE (34–36). Thus, SOCs can be considered channels that are regulated by STIM1 and require the store depletion–mediated clustering of STIM1.
Our laboratory has previously demonstrated that glomerular MCs selectively expressed TRPC1, −3, −4, and −6, among which TRPC1 associated with TRPC4 and −6 (37). Further study demonstrated that TRPC1 mediated Ang II–induced Ca2+ signaling and contraction in this type of cells (38). However, the nature of the TRPC1 channel (store-operated or non–store-operated) and the requirement for binding partners have not been defined. In the present study, we showed evidence that TRPC1 interacted with TRPC4 to form channel complexes mediating SOCE in MCs. STIM1 was required for activation of the SOC, probably via interaction with TRPC4 protein.
Materials and Methods
Mesangial Cell Culture.
Human MCs were purchased from Cambrex Company (East Rutherford, NJ). The procedures and methods for culturing MCs have been described previously (37–40).
Transient Transfection.
Transient transfection was carried out using Lipofectamine and Plus reagents according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Cells were used 24–48 hours after transfection.
Stable Transfection.
Stable transfection was performed to individually or simultaneously knockdown TRPC1 and TRPC4 in MCs (Fig. 3C). Single and double knockdown of the two proteins was carried out by expression of TRPC1 and/or TRPC4 RNAi constructs containing blasticidin and/or hygromycin selectable markers. The antibiotics were added to cell culture medium 48 hours after transfection. Cells surviving after at least 1 week were used for functional experiments.
Quantitative Real-Time RT-PCR.
The total RNA was isolated from MCs using Versagene RNA kit following the manufacturer’s protocol (Gentra System, Inc., Minneapolis, MN). Tagman primers and probes for trpc1, trpc4, trpc6, and β -actin were designed as described in our previous publication (40). RT reaction used 1.0 μ g total RNA, oligo-dT primers, MMLV reverse transcriptase in a final volume of 20 μ l and was incubated at 37° C for 60 min after a denaturation at 70° C for 10 min. The real-time PCR used 2.5 μ l RT product, 0.5 μ M primer, and 0.4 μ M probe, and was performed using ABI Tagman Universal Master Mix in a final volume of 25 μ l. The PCR mix was denatured at 95° C for 10 min, followed by 40 cycles of melting at 95° C for 15 s and elongation at 60° C for 60 s. The assay was run on SmartCycler (Sunnyvale, CA) and fluorescence changes were monitored after each cycle. The average Ct (threshold cycle) of the fluorescence unit was used to analyze the mRNA levels. The trpc mRNA levels were normalized by actin mRNA levels. Quantification was calculated as follows: mRNA levels (percent of control) = 2Δ CT, where Δ CT = CT, TRPC – CT, actin.
Immunoprecipitation and Immunoblotting.
Cell monolayers were collected with PBS then lysed in 1% Triton X-100 buffer containing (in mM) 150 NaCl, 10 Tris-HCl (pH 7.5), 1 EGTA, 0.2 sodium orthovanadate, 0.2 phenylmethylsulfonyl fluoride, 0.5% NP-40, aprotinin (1 μ g/ml), pepstatin (1 μ g/ml), and proteinase inhibitor cocktail (Roche Applied Science, Indianapolis, IN), followed by centrifugation at 6,000 g for 15 min at 4° C. For coimmunoprecipitation (co-IP) experiments, the cell lysates were incubated for 2 h with the primary antibody indicated, followed by immunoprecipitation (IP) for 1 h with 30-μ l slurry of protein G or A (Amersham Biosciences, Piscataway, NJ) in 50 mM Tris-HCl. Immunocomplexes were then washed five times in lysis buffer. The cell lysates (without precipitation, for inputs) and immunoprecipitates (for co-IP) were undertaken into regular Western blotting.
For immunoblotting experiments, all samples were fractionated by 10% SDS-PAGE, transferred to PVDF membranes, and probed with the indicated primary antibodies. Bound antibodies were visualized with Super Signal West Femto Luminol/Enhancer Solution (Pierce Biotechnology, Rockford, IL).
Patch Clamp Procedure.
Conventional cell-attached voltage clamp was carried out as described previously (38, 40). Single-channel analysis was made with a Warner PC-505B amplifier (Warner Instrument Corp., Hamden, CT) and pClamp 9.2 (Axon Instrument, Foster City, CA). The extracellular solution contained (mM): 135 NaCl, 5 KCl, 10 HEPES, 2 MgCl2, 1 CaCl2, and 10 glucose. The pipette solution contained (mM): 135 NaCl, 5 KCl, 1 CaCl2, 3 EGTA, and 10 HEPES. The calculated free Ca2+ concentration in the pipette solution was <10 nM (MTK software). At the time of each experiment, the pipette solution was supplemented with 100 μ M niflumic acid, 10 mM TEA, and 100 nM iberiotoxin to block Ca2+ -activated Cl− channels and K+ channels. Recordings were made at a holding potential of 80 mV (pipette).
Ratiometric Ca2+ Imaging.
[Ca2+ ]i was measured in MCs using dual excitation wavelength fluorescence microscopy with fura-2, as described previously (38, 40). Briefly, MCs grown on coverslips (22 × 22 mm), were loaded with fura-2 by incubation for ~45 min at room temperature in the dark in physiological saline solution (PSS) containing 2 μ M acetoxymethyl ester of fura-2 (fura-2/AM), 0.09 g/dl DMSO, and 0.018 g/dl Pluronic F-127 (Molecular Probes, Eugene, OR). After washout, the coverslip was then mounted to a perfusion chamber (Warner, Model RC-2OH) and placed on the stage of a Nikon Diaphot inverted microscope. Fura-2 fluorescence was monitored by a ratio technique (excitation at 340 and 380 nm, emission at 510 nm) using Metafluor software (Universal Imaging, West Chester, PA). Bath [Ca2+ ] was reduced to less than 10 nM during the experiments by addition of EGTA, according to the Ca2+ concentration program by MTK Software.
Antibodies, Plasmids, and Chemicals.
TRPC1 mouse monoclonal antibody (1F1), HA-TRPC1 expression plasmids, and TRPC1-RNAi construct (shRNA-TRPC1) were obtained from Dr. Leonidas Tsiokas (University of Oklahoma Health Sciences Center, Oklahoma City, OK). RNAi construct for human TRPC4 was kindly provided by Dr. Mitch Villereal (University of Chicago, Chicago, IL). Rabbit anti-TRPC1 antibody (Fig. 6) and rabbit anti-TRPC4 antibody were purchased from Sigma-Aldrich (St. Louis, MO). GOK/STIM1 mouse monoclonal antibody was purchased from BD Biosciences Pharmingen (San Jose, CA). All chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Statistical Analysis.
Data are reported as means ± SE. The one-way ANOVA plus Student-Newman-Keuls test, Student’s unpaired t test, and Student’s paired t test were used to analyze the differences among multiple groups, between two groups, and before and after treatment in the same group, respectively. P < 0.05 was considered statistically significant.
Results
SOC in MCs.
Thapsigargin (TG) was used to activate SOC by passive depletion of internal Ca2+ stores. Consistent with our previous report (10), MCs in culture have spontaneous, but relatively low, channel activity under resting state. After stimulation with 1 μ M TG, single-channel currents increased strikingly (Fig. 1A). The TG-induced channel activation usually onset within 1 min and reached peak about 3 min after treatment. In some cases, multiple levels of the channel opening could be seen in one patch. Because the pipette solution included niflumic acid, TEA, and iberiotoxin, the currents were unlikely mediated by Cl− or K+ channels in MCs (5). The calculated NPO from 11 cells (Fig. 1B) showed that TG treatment significantly raised channel activity (0.039 ± 0.015 vs. 0.083 ± 0.02, before TG vs. after TG, P < 0.05). However, in the presence of 2-aminoethoxydiphenylborane (2-APB, 20 μ M), or La3+ (2 μ M), or MRS1845 (10 μ M), all of which are known SOC inhibitors (10, 41–43), TG was not able to increase membrane currents (Fig. 1A and B). In agreement with our previous report (10), 2 μ M La3+ not only blocked TG-induced response completely, but significantly inhibited basal activity as well.
TRPC1 and TRPC4 Contribute to SOC Activity.
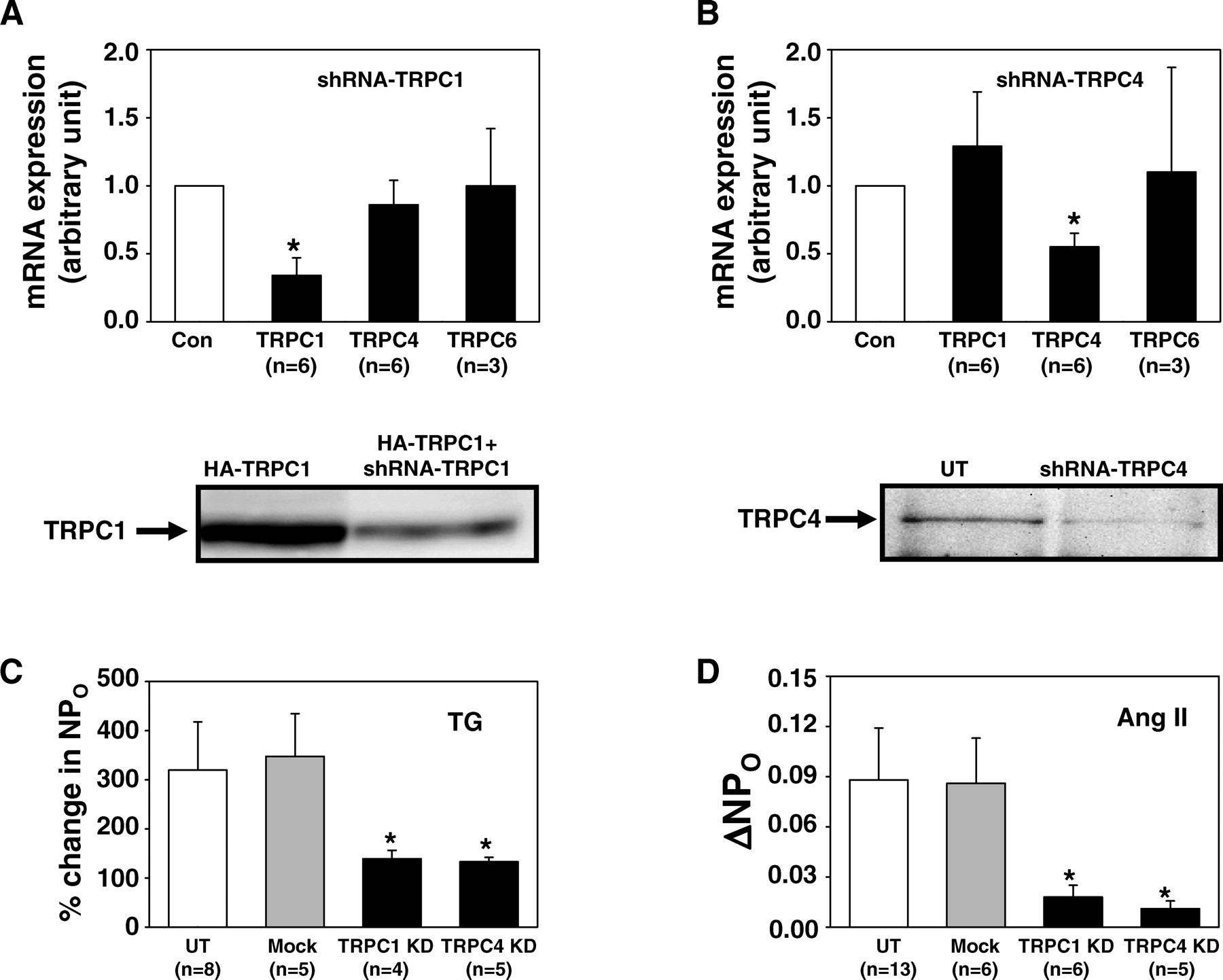
TRPC1 and TRPC4 have been previously identified in MCs in vitro as well as in vivo (37). We then determined if the two TRPC subtypes were components of SOC in MCs by examining the effect of knocking down the individual TRPCs on SOC activity. Knockdown of TRPC1 and TRPC4 was accomplished by transient transfection with RNAi plasmids that produced short hairpin RNAs specifically targeting on human trpc1 (designated as shRNA-TRPC1) or trpc4 (designated as shRNA-TRPC4). GFP expression plasmids were co-transfected with the RNAi constructs or empty vectors (Mock control) at 10-fold less for identification of successfully transfected cells. Quantitative real-time RT-PCR showed that shRNA-TRPC1 significantly reduced the mRNA expression of TRPC1, but not that of TRPC4 or TRPC6, and shRNA-TRPC4 significantly decreased the mRNA expression of TRPC4, but not that of TRPC1 or TRPC6 in human MCs (the upper panels in Fig. 2A and B). Western blot showed that these RNAi constructs efficiently reduced the expression levels of corresponding TRPCs at protein level (the bottom panel in Fig. 2A and B). In the cell-attached patches, we found that either knockdown of TRPC1 or TRPC4 with these constructs significantly inhibited the channel activation induced by TG (Fig. 2C). These results indicated that both TRPC1 and TRPC4 are contributors to SOC in MCs.
We then examined whether TRPC1 and TRPC4 were involved in the physiological signaling pathway for activation of SOC in MCs. Ang II is an important agonist that regulates a variety of physiological processes in MCs (1, 38). Previous study has demonstrated that Ang II activated SOC in MCs (4, 44). Using the cell-attached patch clamp, we found that, similar to the effect on the TG-induced responses, knockdown of either TRPC1 or TRPC4 significantly reduced the channel activity stimulated by Ang II (Fig. 2D). Voltage-operated Ca2+ channel was not responsible for the Ang II–induced response, since the pipette solution was nearly Ca2+ free and contained 1 μ M nifedipine. These data suggest a physiological relevance of both TRPC1- and TRPC4-mediated channel responses in MCs.
TRPC1 and TRPC4 Form Channel Complexes Mediating SOCE.
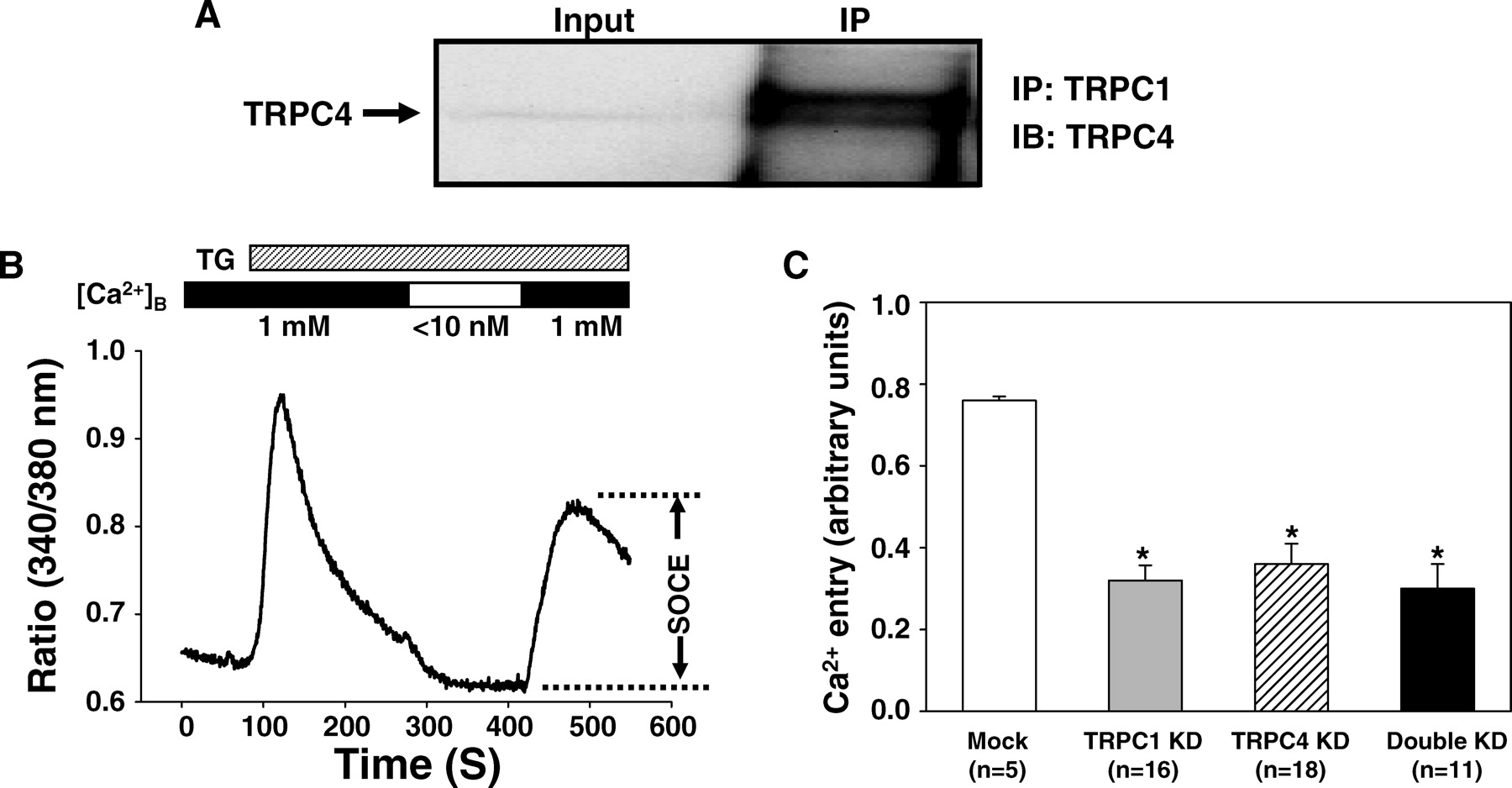
Physiologically, TRPCs are thought to function as channels by forming heteromultimers (13, 21, 22). We speculated that the functional SOCs in MCs might be formed by protein complexes of TRPC1 and TRPC4. This speculation was supported by co-immunoprecipitation, which showed a physical interaction between endogenous TRPC1 and TRPC4 in MCs (Fig. 3A). We further tested this hypothesis by comparing the effects on SOCE by double knockdown of TRPC1 and TRPC4 with that of single knockdown of TRPC1 or TRPC4. Fura-2 fluorescence measurement was used to indicate the changes in [Ca2+ ]i and a standard Ca2+ -readmission protocol was employed to evaluate SOCE (10). In agreement with our previous report (10), under 1 mM Ca2+ extracellular solution, application of 1 μ M TG evoked a rapid and striking cytosolic Ca2+ transient followed by a sustained elevation of [Ca2+ ]i. Removal of Ca2+ from the extracellular solution resulted in a decrease in [Ca2+ ]i and then readmission of Ca2+ into the Ca2+ free bath resulted in a marked rise in [Ca2+ ]i (Fig. 3B), an indicator of SOCE (10). Consistent with the patch clamp data (Fig. 2B), this Ca2+ entry in response to Ca2+ readmission was significantly inhibited by knockdown of single TRPC1 or TRPC4. Double knockdown of both subtypes of TRPC simultaneously had a similar inhibition on SOCE to that induced by single knockdown of either TRPC (Fig. 3B). There was no significant difference between the double knockdown and single knockdown of either TRPCs. These results suggest that TRPC1 and TRPC4 may assemble with each other to form functional SOCs in native MCs.
STIM1 Was Required for SOCE in MCs.
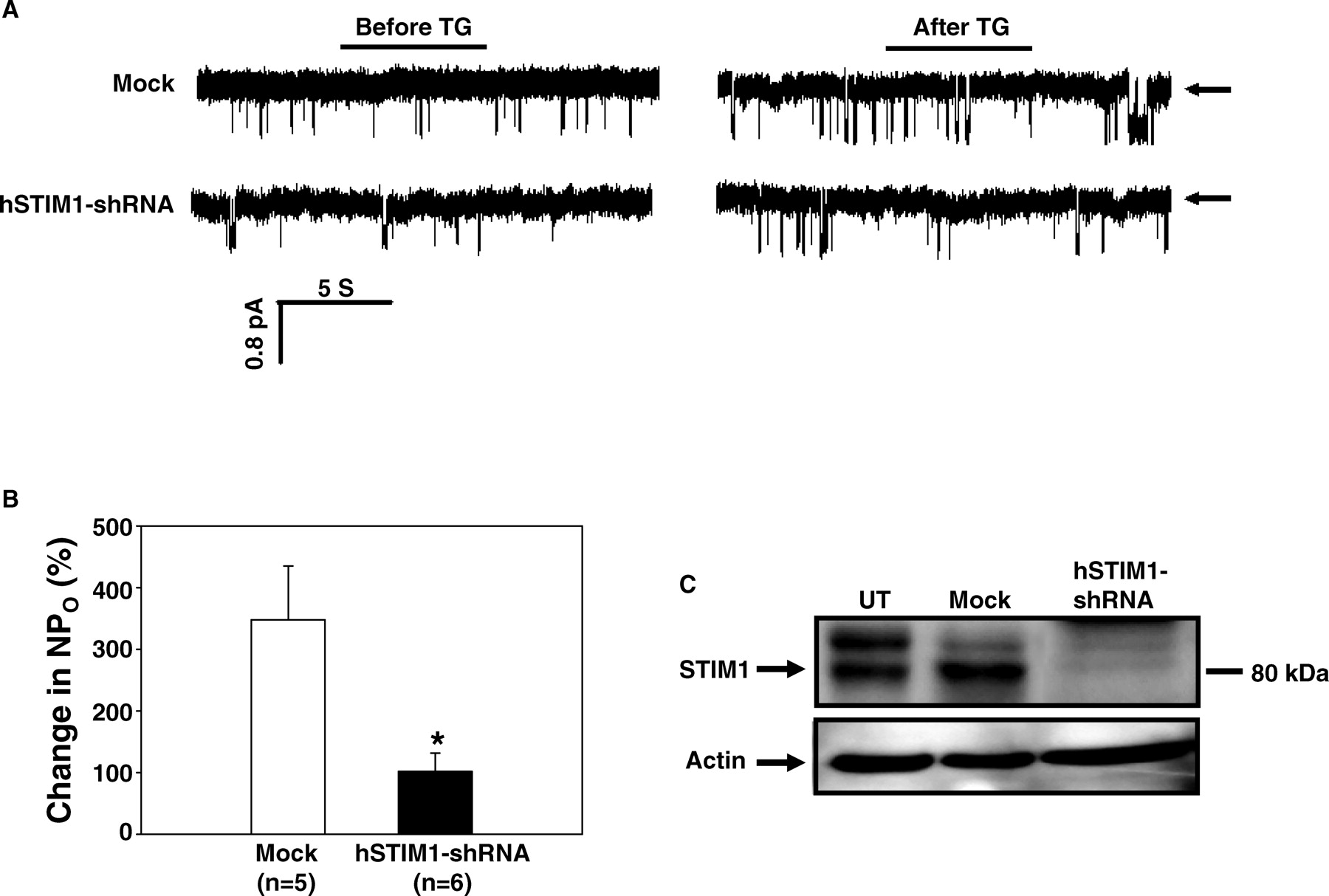
Recent evidence demonstrates that STIM1 acts as a regulator of SOCE by signaling store-depletion in the ER to SOC at the plasma membrane. STIM1 is ubiquitously expressed in a number of cell types, and MCs are no exception. Here, we confirmed a critical role for STIM1 in SOCE in MCs using the cell-attached patch clamp approach. As shown in Figure 4A and B, specific knockdown of STIM1 by transient transfection of specific RNAi constructs (hSTIM1-shRNA) dramatically attenuated TG-stimulated increase of NPO. Western blot verified that the RNAi constructs significantly reduced the expression of STIM1 protein in MCs (Fig. 4C).
STIM1 Was Physically and Functionally Coupled with TRPC1/TRPC4 Complex.
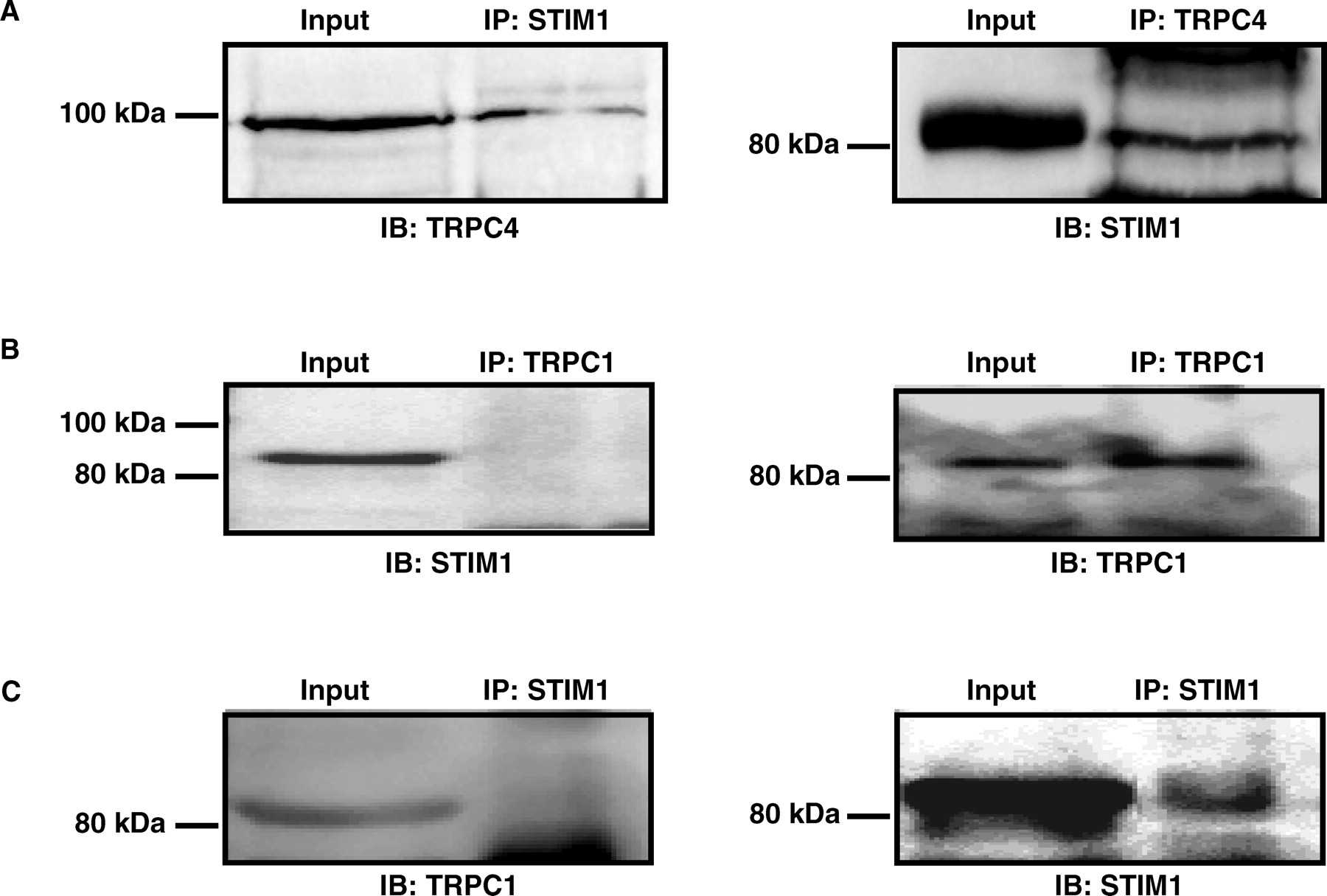
Recent studies suggest that store depletion triggers STIM1 to cluster in a local region of the ER membrane that closely apposes to the plasma membrane in which SOC proteins cluster (34). If TRPC1/TRPC4 complexes are the components of SOC, it is possible that there is a physical interaction between the TRPC proteins and STIM1. We tested this postulation using co-IP assay and found that STIM1 was able to pull-down TRPC4 and vice versa (Fig. 5A). The estimated amount of TRPC4 interacting with STIM1 was ~3% of total TRPC4 protein. Although this is a small percentage, considering the influence from the affinity of antibody and protein beads, and experimental procedures of IP that could cause loss of the protein-protein interaction, these biochemical data provide evidence of a physical interaction between TRPC4 and STIM1. However, we could not resolve a consistent interaction between STIM1 and TRPC1 by co-IP (Fig. 5B and C), suggesting that the TRPC4-STIM1 interaction might be specific in MCs.
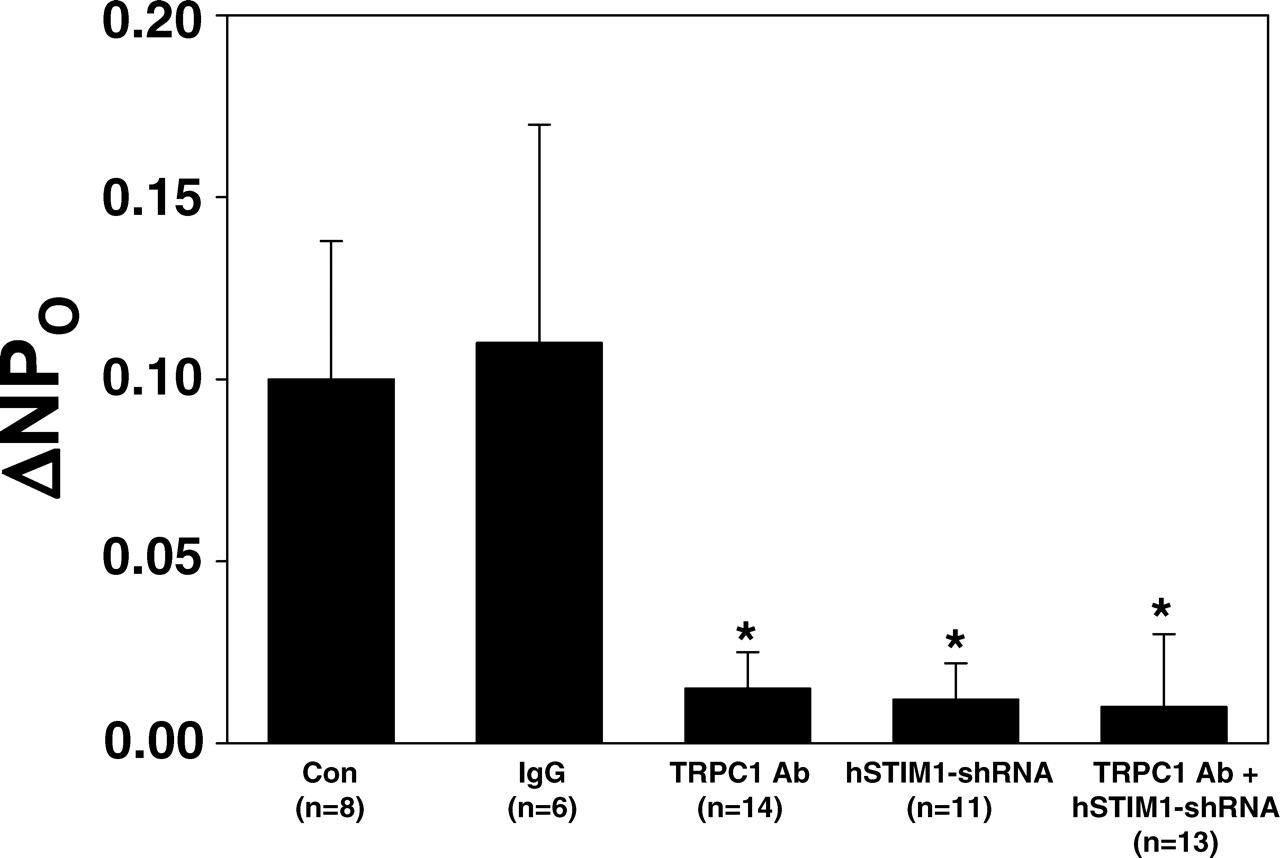
We speculated that if SOC was activated by the interaction between STIM1 and TRPC1/TRPC4 complexes, then the effect on the channel activity should be comparable between inhibition of the individual molecules and of these molecules simultaneously. We took advantage of the TRPC1 antibody (from Sigma) that can specifically block TRPC1 channel (38, 45, 46) and compared the inhibitory effects on SOC activity between individual and simultaneous downregulation of STIM1 and TRPC1. As shown in Figure 6, in the cell-attached patches, either single knockdown of STIM1 or blockade of TRPC1 channel by inclusion of the Sigma TRPC1 antibody (300 ng/ml) into the pipette solution significantly attenuated the TG-induced channel activation. The TRPC1 antibody effect was specific because the same amount of rabbit IgG did not affect the TG response. Furthermore, the inhibitory effects by STIM1 knockdown and TRPC1 inhibition were not statistically different. Simultaneous depression of STIM1 and TRPC1 also resulted in a comparable inhibition with knockdown of STIM1 or inhibition of TRPC1 alone. Combined with the data shown in Figures 2 and 3, these results provided further support to our hypothesis that SOCE is mediated by interaction between STIM1 and TRPC1/TRPC4 complexes in glomerular MCs.
Discussion
We and others have demonstrated that SOC participates in Ca2+ signaling in glomerular MCs (4, 10, 44, 47). This channel mechanism may have physiological as well as pathophysiological significance, such as in diabetic nephropathy (6, 48–51). However, the molecular component of this channel is unknown in MCs. Although recent evidence demonstrates that Orai protein is the long-sought molecular entity of the CRAC channel in hematopoietic cells (26, 28, 31), distinct biophysical properties of SOC in other cell types underlie different molecular channel components (10–12). TRPC proteins, most likely TRPC heteromultimers, remain strong candidates of SOC in a variety of cell types (7). Since the distribution of TRPC subtypes and the abundance of single TRPC proteins are distinct in different types of cells (52–54), the compositions of TRPC complexes mediating SOCE might be cell type specific. Our findings in the current study suggest that TRPC1 and TRPC4 are critical components of SOC in human glomerular MCs. This conclusion is supported by two lines of evidence, i.e., a physical interaction between TRPC1 and TRPC4 (Fig. 3A) and a similar inhibition on SOCE by double knockdown of TRPC1/4 and single knockdown of either TRPC1 or TRPC4 (Fig. 3C). Wang et al. recently reported that TRPC1 and TRPC4 were only two isoforms of TRPC in mouse MCs based on RT-PCR assay and speculated that TRPC4 constituted SOC by forming homotetramers (17). However, the role of TRPC1 in SOCE was not examined in their study. It might be that the native SOC is solely composed of TRPC4 in mouse MCs, but requires both of TRPC1 and TRPC4 in human MCs.
Whether there is any other protein, particularly Orai1, contributing to the native SOC in human MCs is unknown. Interestingly, Liao et al. recently reported that Orai1 physically interacted with TRPC3 and TRPC6, and that Orai1, by interacting with the TRPCs, acts as regulatory subunits that confer store depletion sensitivity to these channels (55). Orai1 is also critical for TRPC1-mediated SOCE in platelets and salivary glands (56, 57). The similar mechanism may also exist in human MCs in which TRPC1/ TRPC4 complexes constitute the pore-forming subunits of SOC and Orai1 acts as an accessory subunit regulating the channel activation.
There has long been a debate as to what mechanism accounts for the communication of store-depletion in the ER to activation of SOC channels at the plasma membrane. Recently, there has been strong evidence for STIM1 as a key component of SOC. In this proposed mechanism, STIM1 acts as a Ca2+ sensor in the ER. Upon store-depletion, STIM1 translocates into punctae directly apposed to the plasma membrane, where it activates SOCE (34, 58). Thus, SOCs could be considered as the channels that are regulated by STIM1 and require the store-depletion–mediated clustering of STIM1. In the current study, we confirmed that STIM1 was required for SOCE in human glomerular MCs, which is in agreement with an earlier report by Zhang et al. in rat MCs (47). We further demonstrated in the present study that the STIM1-dependent SOCE in MCs was mediated by TRPC1/TRPC4 complexes because simultaneous knockdown of STIM1 and blockade of TRPC1 channel resulted in a comparable inhibition on SOC (Fig. 6). Moreover, our results further indicate that the STIM1-dependent SOC activation might be through its interaction with TRPC4 protein in MCs. Indeed, STIM1 has been shown to selectively bind to certain isoforms of TRPC proteins, including TRPC1 and TRPC4 (14, 59). Different from the studies by Yuan et al. and Huang et al. (14, 59), we were not able to demonstrate any physical association between STIM1 and TRPC1. This discrepancy may simply be due to a weak binding between STIM1 and TRPC1 in MCs, or to distinct TRPC1 antibodies (Alomone in their study and 1F1 antibody in our study). Relatively low expression level of TRPC1 in MCs may also make it difficult to be isolated by immunoprecipitation.
In conclusion, the findings from the current study suggest that in human glomerular MCs, the TRPC1/TRPC4 complexes constitute the functional subunits of SOC and are activated through interaction between STIM1 and TRPC4. How STIM1-TRPC4 interaction is regulated upon store depletion remains unclear, and further study is needed to explore the detailed mechanism. Given a significant role of MCs in the regulation of glomerular filtration, identification of Ca2+ signaling pathway and its regulatory mechanisms, such as SOCE, in MCs has important physiological and pathophysiological relevance.
Co-immunoprecipitation, showing STIM1 interaction with TRPC4, but not TRPC1. Cell-attached patch clamp experiment, showing the effect on TG-induced channel activity by various treatments in MCs. Δ NPO, the difference in NPO before and after TG treatment; Con, control; IgG, rabbit immunoglobulin; TRPC1 Ab, TRPC1 antibody; hSTIM1-shRNA, short hairpin RNAi construct for human STIM1; N, number of cells analyzed. * Significant difference compared with both Con and IgG group.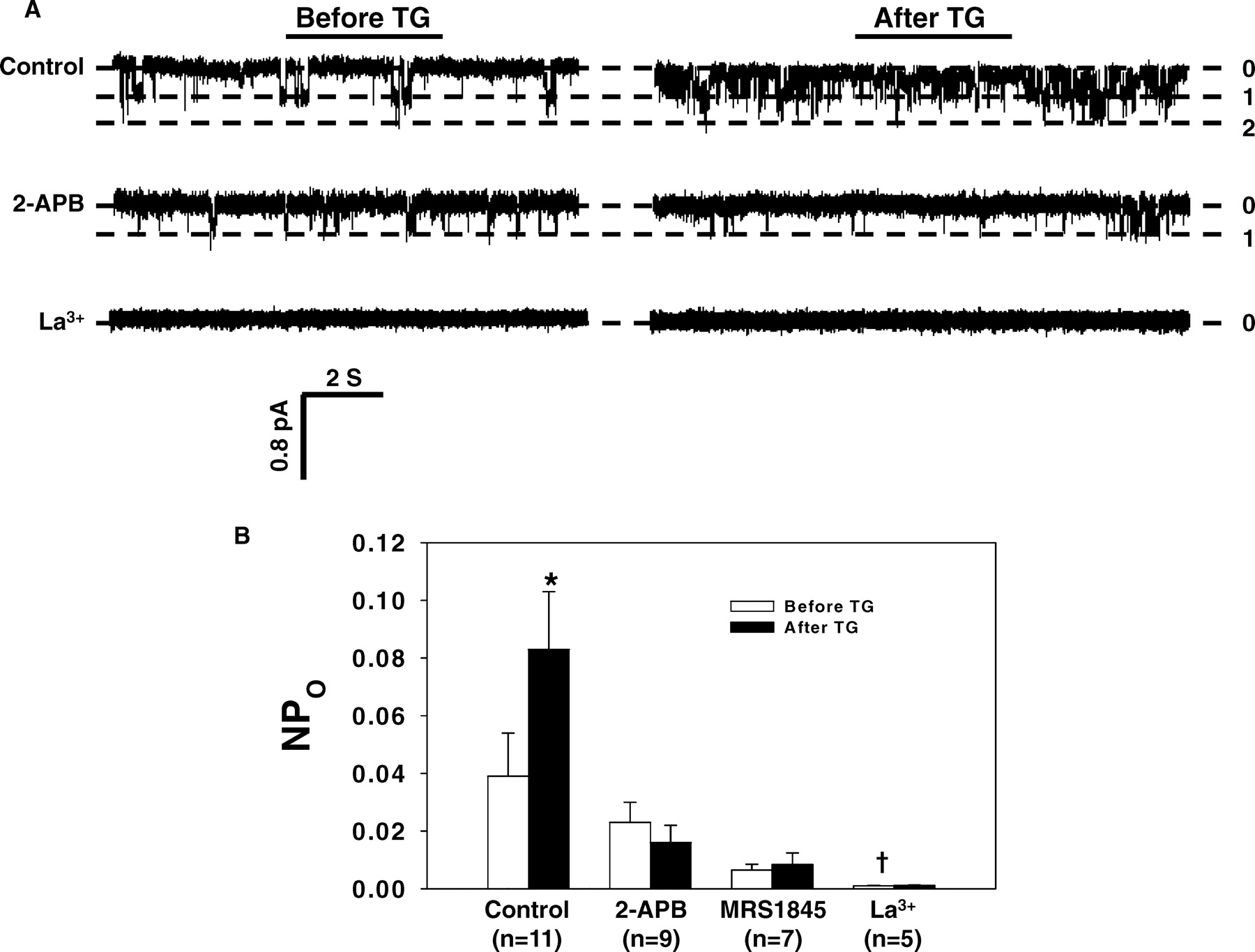
Footnotes
This study was supported by Young Investigator Grant from National Kidney Foundation (Ma) and Research Award from American Diabetes Association (Ma).
Acknowledgements
We thank Dr. Leonidas Tsiokas (University of Oklahoma Health Sciences Center, Oklahoma City, OK) for generously providing us with human TRPC1 expression plasmids, shRNA-TRPC1 constructs, and 1F1 TRPC1 antibody. We also thank Dr. Mitch Villereal (University of Chicago, Chicago, IL) for kindly offering us shRNA-TRPC4 constructs.