
Abstract
The rapid growth of malicious software (malware) production in recent decades and the increasing number of threats posed by malware to network environments, such as the Internet and intelligent environments, emphasize the need for more research on the security of computer networks in information security and digital forensics. The method presented in this study identifies “species” of malware families, which are more sophisticated, obfuscated, and structurally diverse. We propose a hybrid technique combining aspects of signature detection with machine learning based methods to classify malware families. The method is carried out by utilizing Profile Hidden Markov Models (PHMMs) on the behavioral characteristics of malware species. This paper explains the process of modeling and training an advanced PHMM using sequences obtained from the extraction of each malware family’s paramount features, and the canonical sequences created in the process of Multiple Sequence Alignment (MSA) production. Due to the fact that not all parts of a file are malicious, the goal is to distinguish the malicious portions from the benign ones and place more emphasis on them in order to increase the likelihood of malware detection by having the least impact from the benign portions. Based on “consensus sequences”, the experimental results show that our proposed approach outperforms other HMM-based techniques even when limited training data is available. All supplementary materials including the code, datasets, and a complete list of results are available for public access on the Web.
Introduction
Currently, we can observe a tremendous increase in the use of computers in everyday life and the Internet is playing a great role in people’s daily routines by providing important services. The dramatic advances in information technology and electronic commerce have provided new opportunities for business environments but has also brought about issues of security and confidentiality of information in cyberspace. Since Frederick Cohen used the phrase “computer virus” in 1983, a large number of new malicious codes have emerged every year.
Malicious software also known as malware, in general, refers to all unwanted computer programs such as computer viruses, Trojans, rootkits, worms, adware, spyware, and ransomware [1, 2, 3, 4, 5]. Malware detection is the first step in protecting computer systems. The general way to deal with malware is using anti-virus software, but these programs mostly use signature-based detection techniques and are not capable of identifying new malware and the zero-day threats. Today’s malware techniques are more insidious and better written than before. Some types of malware are more difficult to detect compared to the old malware generations; recent malware generations (i.e., polymorphic and metamorphic) use encryption and obfuscation techniques [1, 6]. They produce different strains of themselves while keeping the inherent functionality the same in order to create an entirely new variant, which is hardly similar to the original variant after reproduction.
Generally, malware detection techniques consist of two main steps. The first one is the malware analysis and feature extraction and the second is the identification phase. For the first step, the two common methods, static and dynamic analysis as well as new hybrid approaches are described in Section 2. The classification methods in the second step are categorized as signature-based detection, behavior-based detection, and statistical-based detection. They are discussed in Section 3.
Our work has been carried out in the field of cybersecurity, in which we focused on malware detection. Given the fact that one aspect of systems operation is time, its production is a sequence of data. Such sequences can be dealt with using process models and profiling approaches. In the case of process modeling, profiling and its related issues, hidden Markov models and in particular profile hidden Markov models are considered to be powerful tools. They have many use cases in computer science, including almost all issues in this field that are arranged by events or have a function based on time. Audio processing and speech recognition, handwriting and text recognition, detection of cyber-attacks and network penetration, biomarker modeling or protein structure prediction, detection and prevention of fraud or extraction of customer behavior patterns in bank transactions, and forecasting financial markets and stock prices are some cases that can be mentioned. HMM-based models have always been highlighted and played a significant role in the proper solutions to such issues. Therefore, modifying and upgrading the HMM-based models, which result in better performance and higher detection and classification accuracy, would have a significant impact on such applications and motif problems that were briefly mentioned above.
The specific sequence-based architecture of HMM decreases the complexity, making it plausible to reduce the overall run-time of the training phase. Furthermore, a plain structure implies that the associated model has a few parameters and thus can be trained on a small amounts of data. Such HMM architectures can lead to apt models if they proportion the structure of the application elegantly. Profile HMM (also known as linear HMM) is one example of these architectures, which is befitted as a motif representation for interesting positions in sequences. It is useful for two reasons. First, its structure is linear to match that of a position and second, it strives to return the process of evolution. The foundation of a profile HMM is a sequence of states, so-called “match states”, which depict the canonical sequence for the family under consideration. Each of the match states corresponds to one position in the canonical sequence. The series of states is similar to a position weight matrix (known as a profile) because each state has a frequency distribution across the whole symbols. Profile HMM also consists of two additional types of states (insert and delete states) in order to model the evolution process. Each delete state stands alongside its corresponding match state, which allows the match state to be skipped. Insert states with self-loops are placed between match states, allowing one or more bases to be inserted between two match states.
Contributions
We develop a statistical model originally introduced by Attaluri et al. [1] to strike obfuscated malware detection and propose a novel profiling algorithm to consider important features of data. The central objective of this paper is to model an advanced profile hidden Markov model (APHMM), which improves the performance of the proposed model. Moreover, we build a model incorporating statistical learning methods for malware behavior analysis using limited data and modify it to deal with binary features while maintaining the high detection rates of malicious activities.
The rest of this work is organized as follows. In Section 2, we review related work. Section 3 presents a brief history of malware detection techniques. In Section 4, the proposed approach and experimental results are described. We discuss our experiments and the utilized datasets in Section 5. The paper concludes with Section 6 where we discuss possible future work.
Related work
As mentioned earlier in Section 1, malware analysis techniques can be divided into two main categories of static and dynamic analysis methods. Static analysis is more popular because it analyzes the structure of malicious code and detects malware without running it in contrast to dynamic analysis, which usually requires emulation and malicious code analysis in runtime. Damodaran et al. identified three general approaches in malware analysis, namely, static analysis, dynamic analysis, and hybrid approaches [7]. Certain malware detection methods are based on static analysis discussed in [1, 3, 4, 5, 6, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18] and only rely on the features extracted from malware or benign files without executing them. On the other hand, dynamic analysis discussed in [5, 19, 20, 21, 22, 23, 24, 25, 26, 27], in which there is runtime monitoring, has been successfully used for malware detection. Recently, hybrid approaches in [7, 28, 29, 30] have been investigated, where static and dynamic features are used for precise identification. In the following sections, previous work is reviewed based on whether they rely on static, dynamic or hybrid static-dynamic analysis.
Static analysis
Static analysis of a malware is performed without the need for program execution. Some useful information we can extract using static analysis is the sequence of operational codes (known as opcodes), and control flow graphs. Khalilian et al. proposed an expert system for static detection of metamorphic malware. Their proposed method, named G3MD, applies graph mining to the opcode graphs of the metamorphic malware family to extract the frequent sub-graphs. Using these sub-graphs, a classifier is learned to distinguish between the benign and malicious files [31]. Christodorescu et al. presented a malware detection technique relying on static analysis and control flow graphs [4]. The approach focuses on recognizing obfuscation patterns in malicious codes and their results with good accuracy. Principal Component Analysis (PCA) [15] was used by Deshpande et al., and Support Vector Machines (SVM) [3] were utilized for malware classification by Singh et al. Fan et al. proposed a sequence mining algorithm to discover malicious sequential patterns along with All-Nearest-Neighbor (ANN) classifier which is constructed based on the discovered patterns for malware detection [32]. Annachhatre et al. hired a clustering algorithm based on features taken from static analysis for malware recognition [8]. Deshpande et al. used function call graph analysis for detecting malicious software [17] while Shanmugam et al. analyzed opcode-based similarity scores which rely on substitution cryptanalysis manners [16]. API call and operational code sequences were employed by Shankarapani et al. to assign whether a part of the code is similar to some specific malicious software [18]. Wagner et al. designed and developed, a knowledge-assisted visual malware analysis system (KAMAS) [33] for behavior-based malware analysis. They focused on a subset of function calls – the critical API calls that can be used for accessing private data and utilizing system resources [33]. In [34], authors developed an assembly code clone search system called ScalClone whose aim is to identify the code clones of the target malware.
Dynamic analysis
In dynamic analysis, the input sample program is executed. It is usually carried out in an isolated environment such as Virtual machines [35] or tools which are famous as sandboxes [36]. Some of the information that can be prepared by dynamic analysis is API calls, system calls, instruction traces, registry changes, and memory writes [7]. Kolbitsch et al. constructed fine-grained models which consider the malware’s behavior based on system calls [37]. These models’ results are depicted in the form of graphs, where the vertices signify system calls and the edges signify affiliation between the calls. Ahmed et al. presented a runtime monitoring tool that obtains statistical features based on spatiotemporal information in the logs of API calls [27]. Eskandari et al. extracted programs’ API calls and then combined them with control flow graphs to build an API-CFG model [23]. In an improved version [30], n-gram methods were used to extract the API calls. Qiao et al. analyzed malware based on frequency analysis of API call sequences [21]. Anderson et al. introduced a malware detection method that uses instruction trace executables logs. The traces are analyzed as graphs, where the nodes of the graphs indicate instructions, and statistics from instruction traces are used for computing transition probabilities [24].
Hybrid approach
Hybrid methods use both static and dynamic analysis aspects. In this section, we review some recent work of this type. Choi et al. introduced a framework for classifying malware using static analysis along with dynamic analysis. They extract malware features using a method called Malware DNA (Mal-DNA) [29]. The main focus of this method is behavior monitoring which occurs in the extraction of dynamic features. Eskandari et al. developed a tool called the HDM analyzer [30]. This tool applies both static and dynamic analysis during a learning phase while the test phase only relies on static analysis. The aim is to benefit from the apparently superior dynamic analysis in the training phase while the static analysis efficiency advantage is maintained at the scoring stage. For comparison, they show that the HDM analyzer has better overall precision and complexity than other static and dynamic analysis methods. The dynamic analysis was employed by Eskandari et al. which was based on the extracted API call sequences. Kabakus et al. introduced a novel hybrid Android malware analysis approach namely mad4a which uses the advantages of both static and dynamic analysis techniques and finds some unknown characteristics of Android malware through the used hybrid approaches [38].
Background
In this section, we present a brief overview on malware detection techniques and explain the general concepts used in this paper, paying special attention to profile hidden Markov models, which are the basis for our performance evaluation. We also review some notable previous works.
Malware detection
Various advanced recognition techniques have been considered in recent studies on malware detection. The common methods used in this field are: signature-based approaches such as pattern matching [4] and N-gram based [39] techniques; behavior-based (also called heuristic-based) approaches such as control flow graph [4], support vector machines [3], decision forest, K-means, and phylogenetic-tree; and statistical-based approaches such as hidden Markov models [11] and behavior profiling with hidden Markov models (also known as profile hidden Markov models) [1, 40]. We briefly review these methods before explaining PHMMs in detail.
Signature-based detection
Signature-based detection is extensively used in anti-virus programs [2, 3, 4]. A signature is a sequence of bytes, which is used to recognize malware belonging to a certain family. An anti-virus program which works based on the signature-based analysis must hold the signature repositories of the known malicious software. These databases are regularly updated by detecting and adding new malicious programs. This approach is efficient and effective for all known types of malware. A disadvantage of this approach is that it must have an up-to-date database and if a piece of malware is not present in the database, it will not be detected. In addition, simple obfuscation techniques are able to defeat and evade this method [1, 5, 7, 32, 41].
Behavior-based detection
Behavior-based detection focuses on transactions with the operating system while malware is being executed. The behavior of malware/benign files is analyzed in a training step. Based on patterns taken during the training phase, an input file is divided into malware or benign in the test step [33, 42]. In this method, the activities of a file are monitored to identify the functional patterns of the object and then to indicate whether the input file is malicious, or at least suspicious without scanning the file to determine its digital signature.
Statistical-based detection
Some techniques such as Markov process models used for malware detection are based on statistical analysis of features taken from the malware file. For example, PHMM was used by Attaluri et al. for identifying sophistical malware [1]. This technique has been used as a benchmark in other studies [1, 10, 11, 19, 20, 40, 43]. PHMMs have also been used in Bioinformatics to represent families of proteins [44]. Protein sequences belonging to one family are aligned to construct a multiple sequence alignment, which is used as the family’s representative profile hidden Markov model. These profiles can be further used to classify new proteins. Due to the inherent similarity of the production of metamorphic malware to the evolution and mutation process in the gene or protein sequences, PHMM is proposed as a potential statistical model for classifying malware families.
Markov chains
Assume that there are several states with transition probabilities for each move from one state to another. Markov chain rule states that each state is independent from all the other states. However, if we make each state dependent on its previous state, the first-order Markov chain will be established. In higher order Markov chains, more previous states are taken into account in the process of computing the probability of one state. In other words, in the
Markov chain for DNA.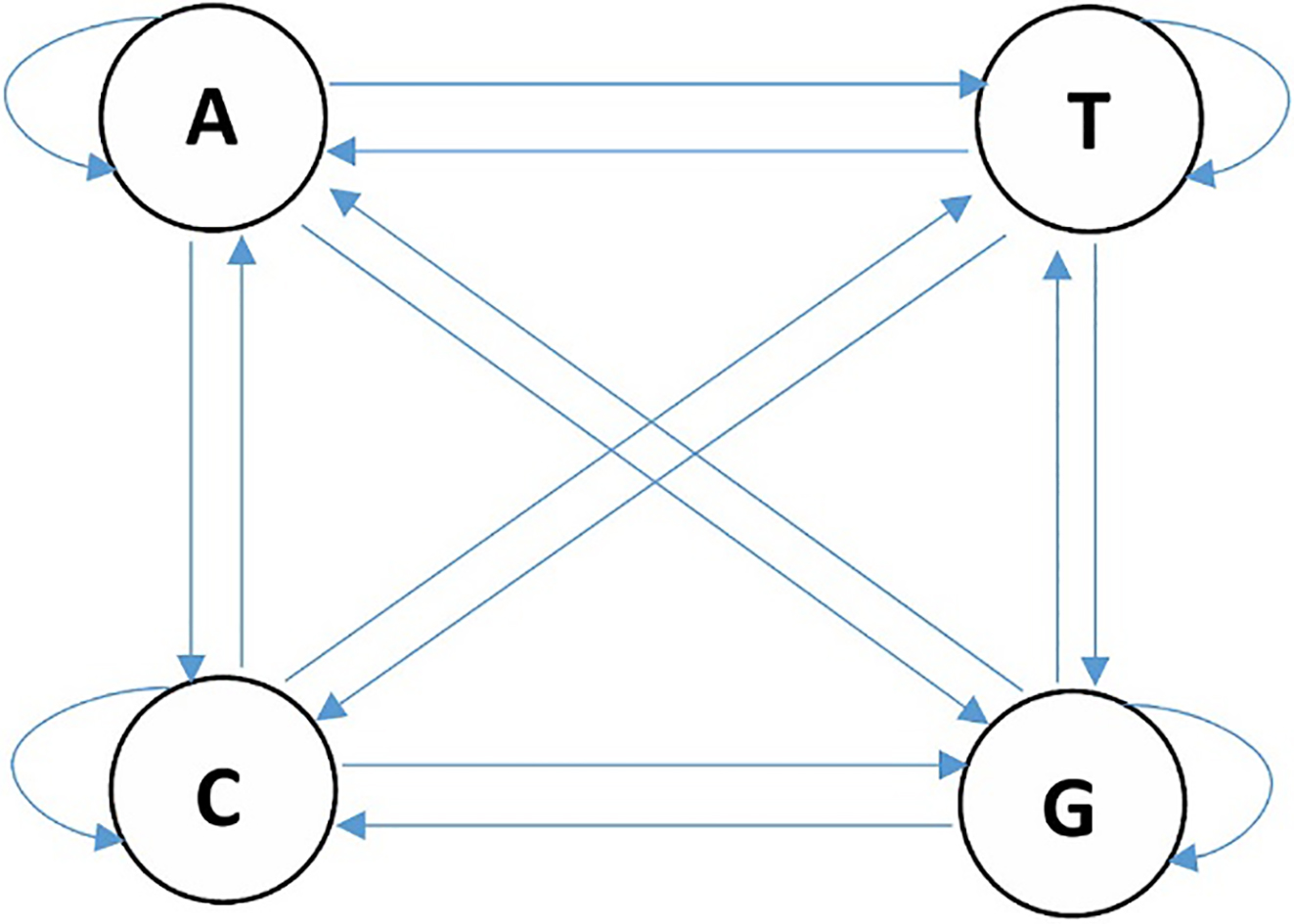
As described in Section 3.2, in higher order Markov chains each state depends on more than one previous state. The higher order Markov chains can also be represented as first-order ones by combining the states and the symbols. For example, a second-order Markov chain with a four-symbol alphabet such as {A, C, T, G} for representing a DNA sequence such as ATCCGA can be represented as a five-state first-order Markov model with states AT, TC, CC, CG, GA. This type of representation is called n-gram (bigram in this example). An n-gram is a commonly used concept in natural language processing, which we are able to use for detecting malware [39] as a data segmentation technique during pre-processing in order to perform feature extraction as has been carried out by Kang et al.
Hidden Markov models
A hidden Markov model (HMM) is a statistical approach for modeling systems which are assumed to contain Markov processes with hidden states and is based on the discrete hill climbing method [45]. HMMs have a variety applications in artificial intelligence area such as speech recognition, computational molecular biology, and malware detection [46]. An HMM can be made up of a series of observations which are affiliated to the “hidden” Markov process states (orange circles in Fig. 2) by a set of discrete probability distributions [7].
Generic hidden Markov model example [7].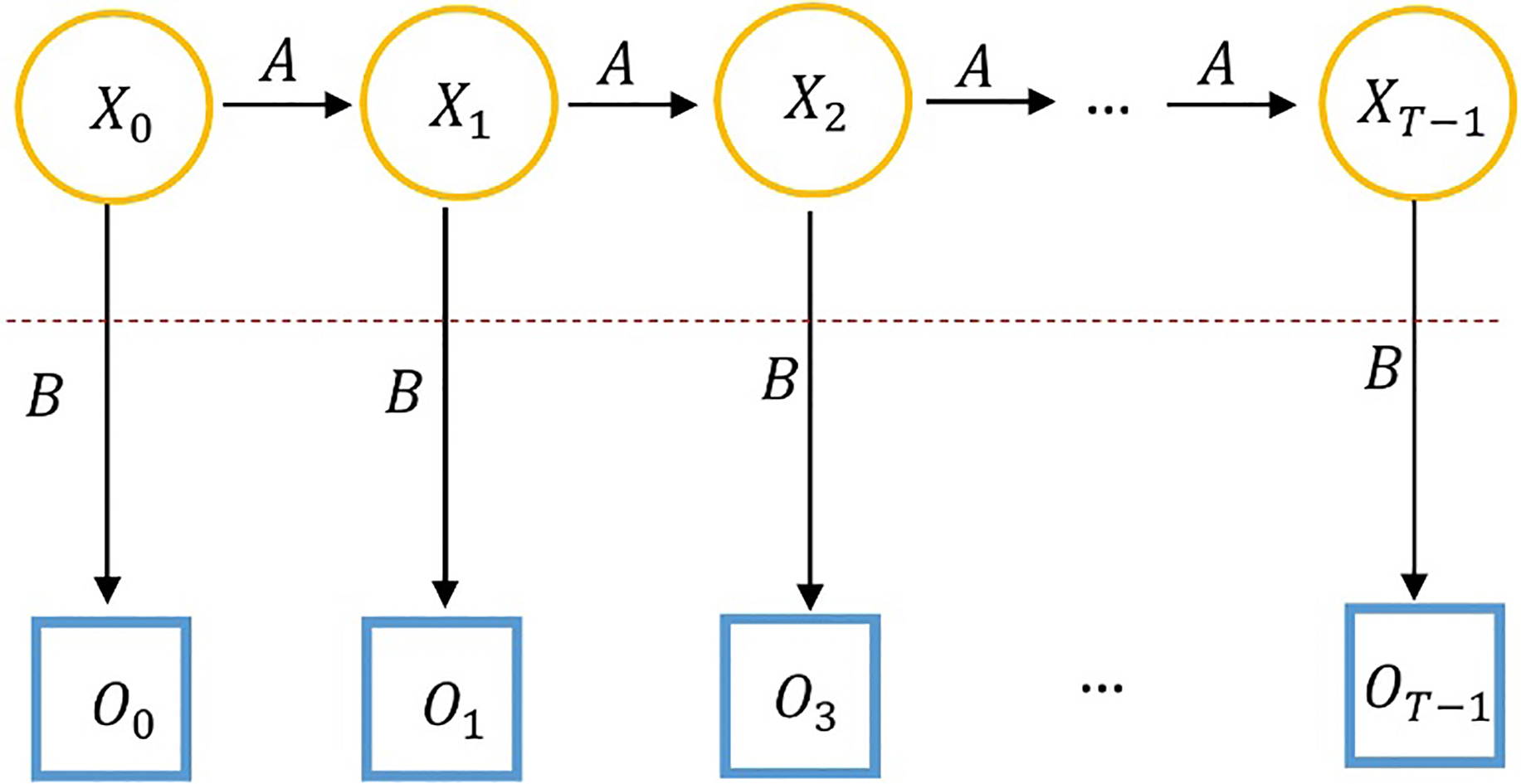
The practical advantage of HMMs is in solving each of the following three problems [45].
Problem 1: Evaluating
“Given an HMM
Problem 2: Decoding
“Given an HMM and an observation sequence
Problem 3: Training
“Given an observed sequence
In this paper, our focus is on the third problem; our main goal is to train a model based on observation sequences extracted from a given malware family. Then, in order to make decisions about unknown files, the solution for problem 1 is utilized for scoring the observation sequences which are extracted from malware files. The sequences extracted from benign files are also scored the same way. A complete discussion of the solutions for HMM’s three problems can be found in [45]; reference [46] is also a valuable one to find additional information on them. In addition, reference [41] contains some information about the application of HMMs in malware detection.
Profile hidden Markov models can be used to classify malicious files into families or groups based on their behavior on the host system. PHMMs have been widely used in the field of bioinformatics to find the similar proteins and DNA sequences in a large database [43, 44]. As is presented in Fig. 3, a PHMM consists of three types of states: matches (rectangles), inserts (ovals) and deletes (circles). Using this particular model helps us overcome the obstacles caused by polymorphic and metamorphic malware. This model can lead to a high degree of classification accuracy compared to other advanced methods in this field. PHMM is a special case of standard hidden Markov model that is designed to cope with fundamental problems in bioinformatics.
A Profile Hidden Markov Model (PHMM) with three match states.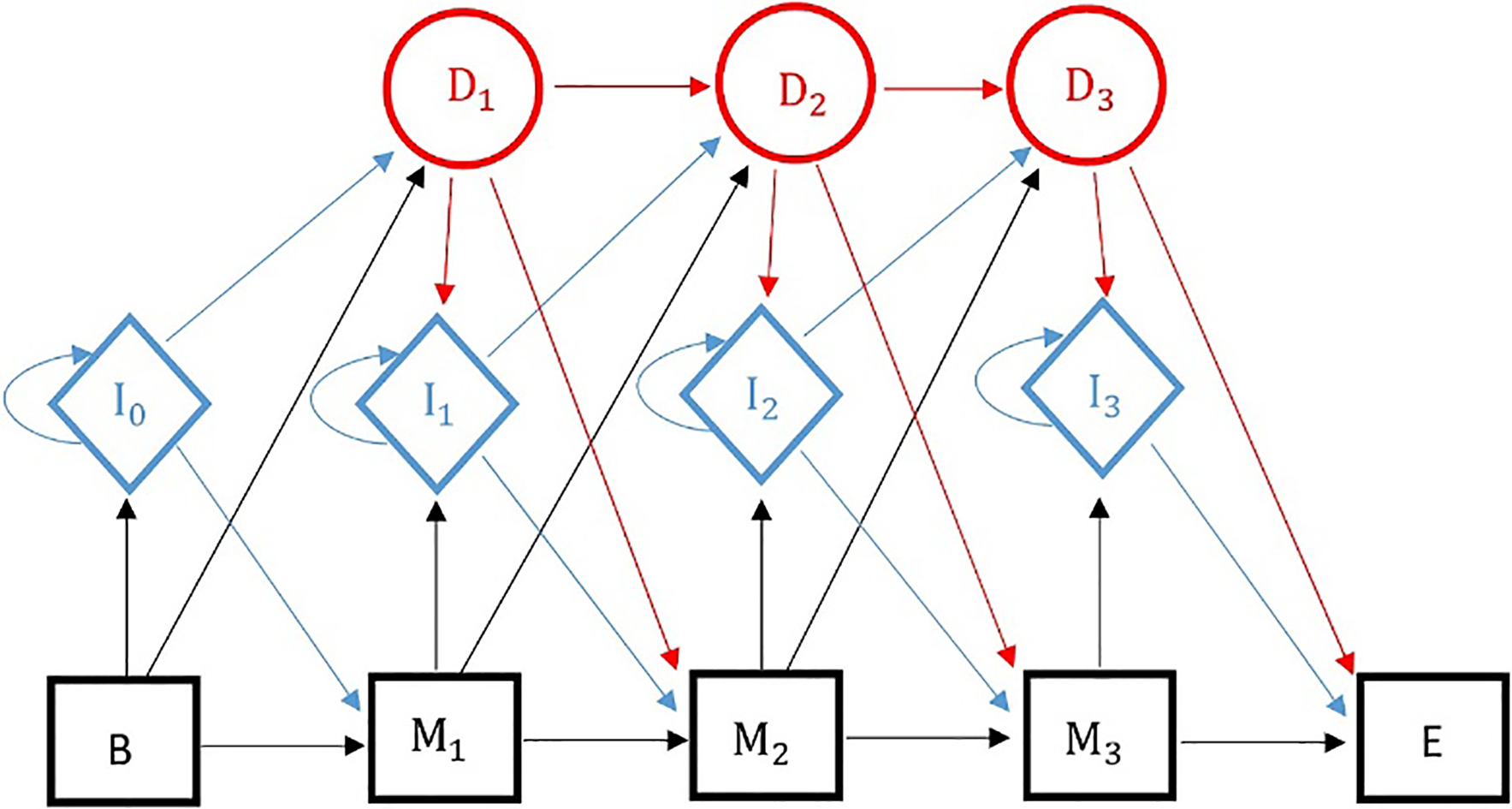
One key step in the PHMM construction procedure is the multiple sequence alignment. A similarity score can be computed for the MSA of DNA sequences which can be used to determine whether the gene sequences are descended from a common ancestor. In malware detection, in order to decide whether the unknown file of the input, which is a sequence of symbols, belongs to a malware family, the file is presented to the models built from MSAs produced from each malware family. Each malware family is modeled by a PHMM and the Viterbi algorithm for PHMMs can be used in this step to compute the similarity score of the sequence to each PHMM model.
Partial order alignment
Partial order alignment (POA) is a fast technique for multiple sequence alignment in bioinformatics. POA guarantees a desirable alignment for each new sequence, as compared to any sequence in the MSA. POA is also scalable and is capable of managing INDELs or branching in the alignment phase. Figure 4 shows an example of how POA constructs a directed acyclic graph for aligning two sequences. This algorithm makes it possible to build MSA for complex structures [47, 48, 49].
Partial order alignment (POA) [50].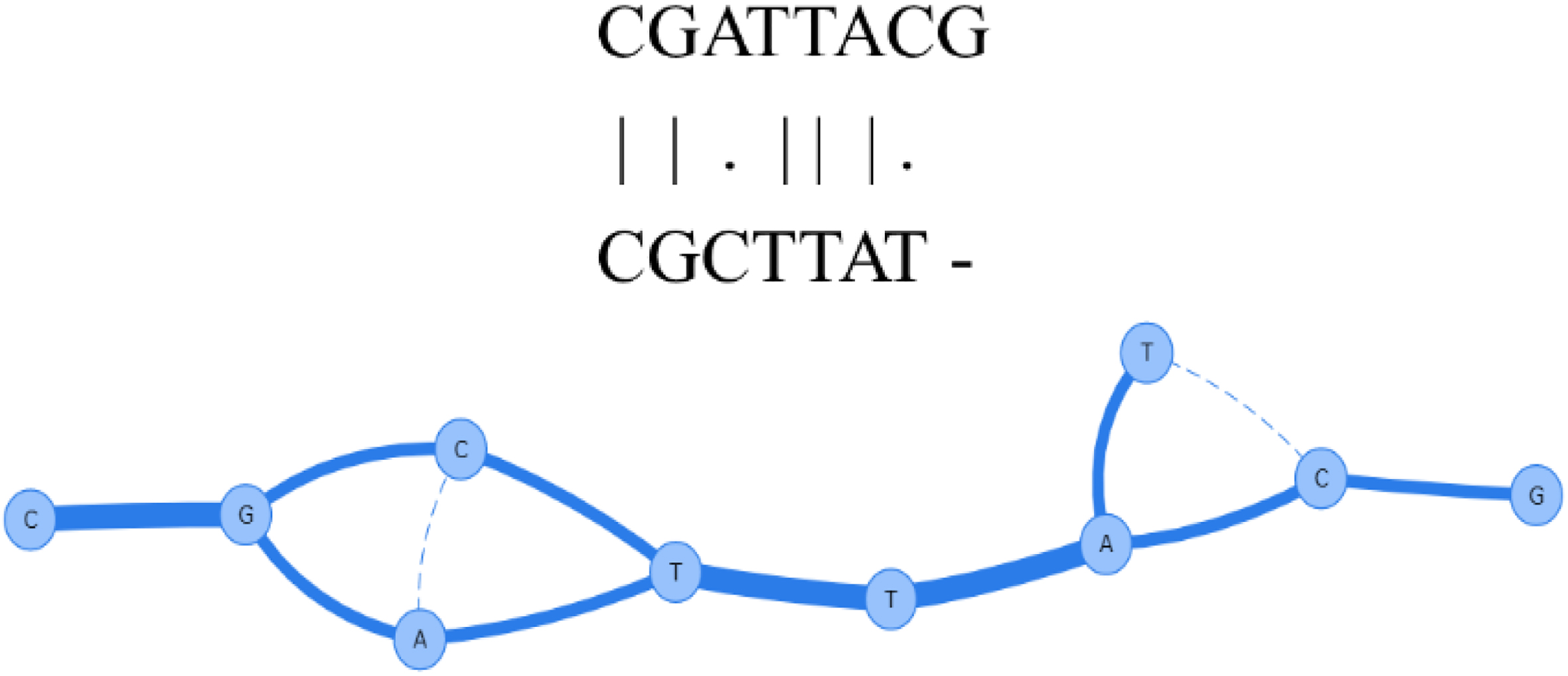
Partial order alignment is a graph-based algorithm that acts the same way as a dynamic programming method does. The only difference between the original dynamic programming algorithm (i.e., Needleman-Wunsch Algorithm; Fig. 5) and POA is that instead of the two sequences used in the former, a graph and a sequence are used as the input of the latter.
The original dynamic programming algorithm [50].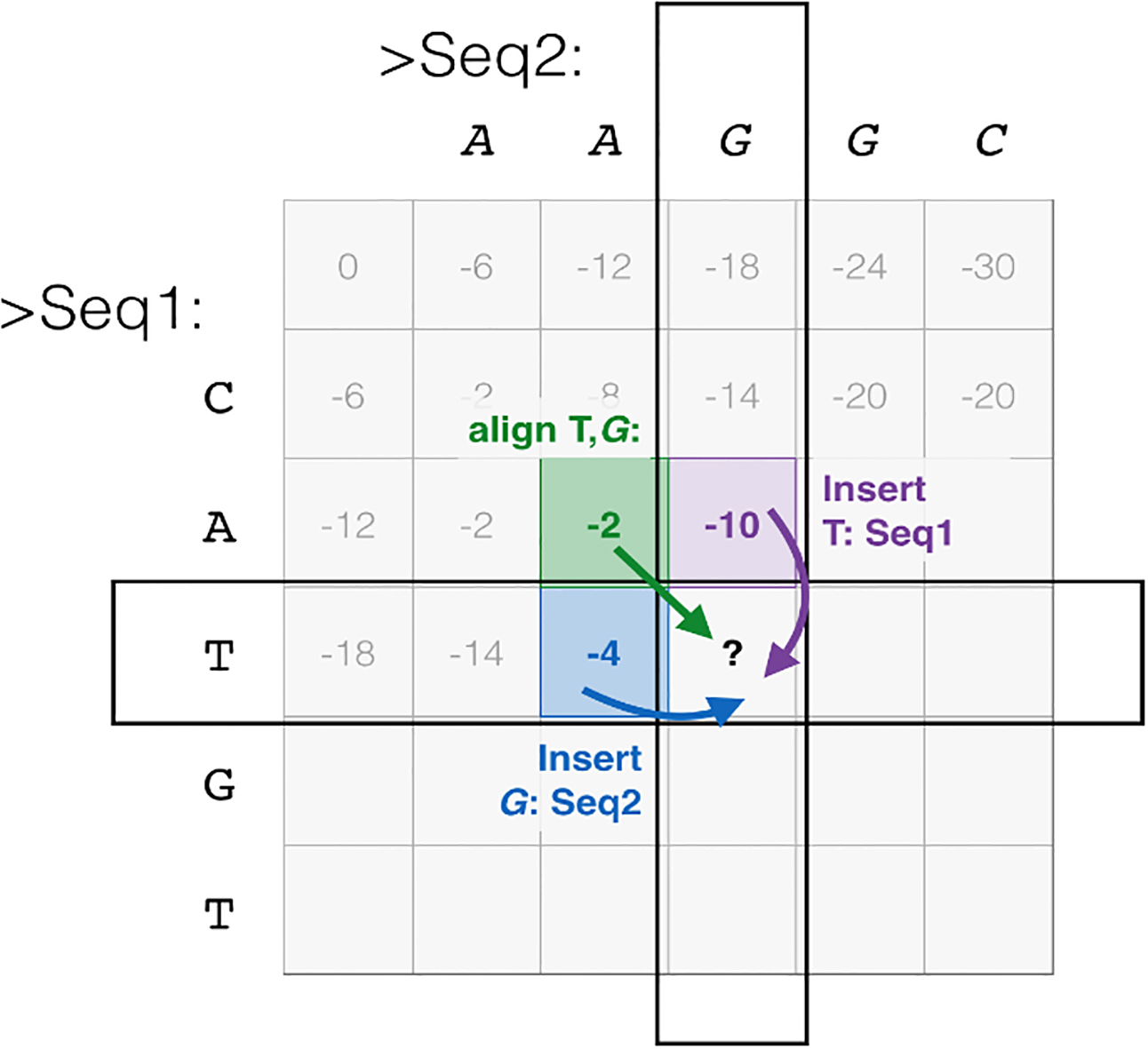
For each alignment, the generated weighted DAG is updated and topologically ordered. Figure 6 depicts one step in updating the directed graph by POA. Finally, the consensus sequences made from this graph can be used to build an advanced PHMM.
String to graph alignment [50].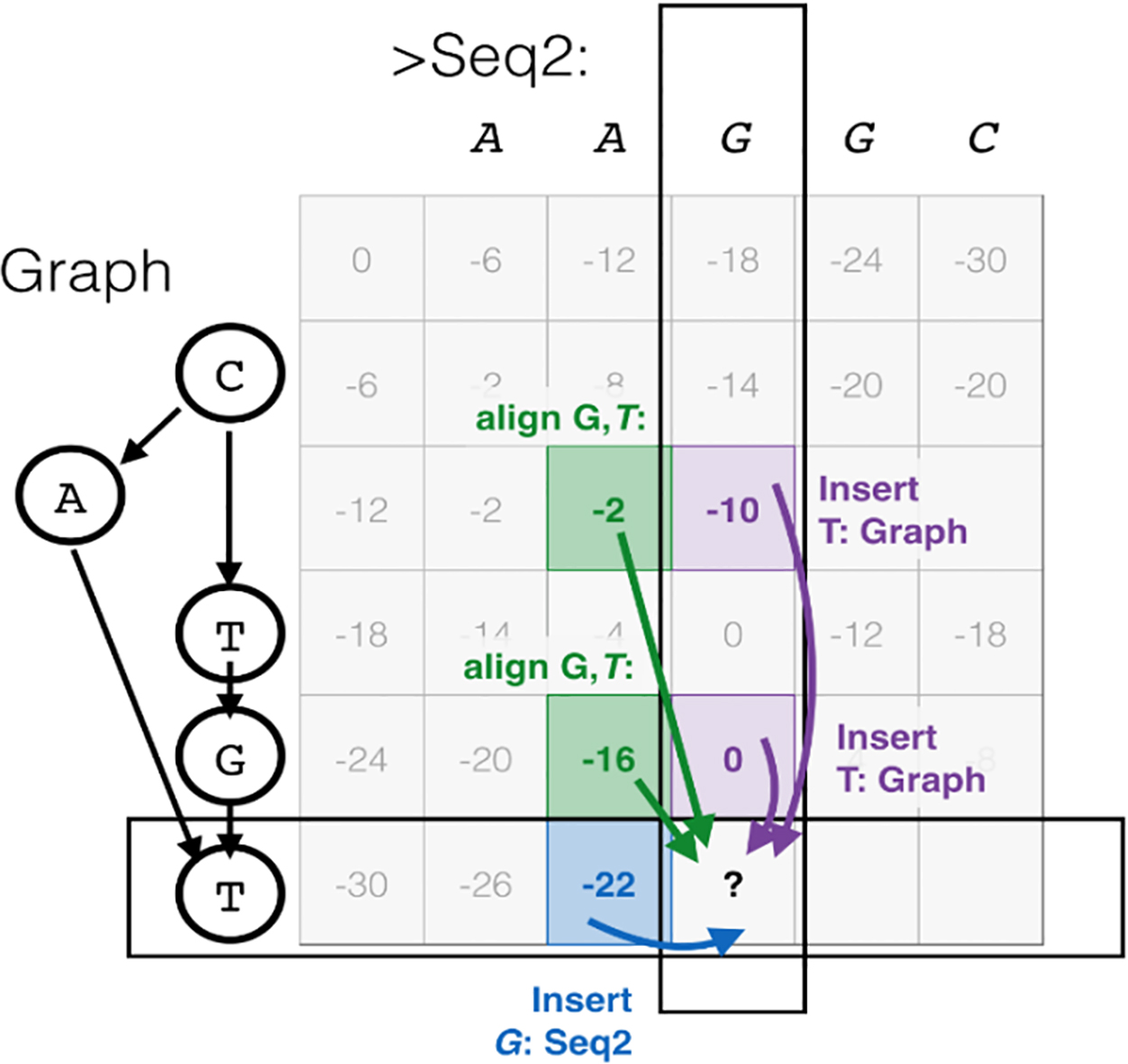
There are two main challenges in using machine learning techniques for malware detection problem. The first one is to develop the machine learning algorithm which is addressed at the training phase of our proposed method and the second challenge is to identify and select an appropriate feature set. The consensus sequences (or canonical sequences) are produced in the process of generating multiple sequence alignments. A consensus sequence is a sequence which is constructed from the weighted DAG generated by POA algorithm and is a representative of all the sequences in the MSA (Fig. 7). A PHMM may have more than one consensus sequence if the sequence family members are diverse.
An example MSA along with the consensus sequence (in the red box).
As is shown in Fig. 7, the consensus sequence is added to the constructed MSA in order to obtain an Advanced Profile Hidden Markov Model (APHMM). Therefore, the features related to each family are emphasized leading to improvement in the performance of our statistical model. Note that the proposed model is superior to standard PHMMs in terms of improved MSA used in building the APHMM, and the model structure is unaltered. The second challenge has recently been the focus of many studies and the existing techniques have produced acceptable results. Our model selection phase is equivalent to scoring every opcode sequence against the constructed PHMM belonging to each malware family. Detection and recognition of malware with high accuracy and speed is one of the most important goals of researchers in the computer security field. To identify malicious code, our proposed approach is based on static behavior and statistical pattern recognition. In this method, we use PHMMs to identify the families of malicious software. By modifying the way of creating MSA, which is considered as learning step in this model, we are able to achieve desirable results. In addition to 100% accuracy, low false positive rate, simplicity, and low computational overhead are other outcomes of the proposed method.
For training and scoring, we used opcode sequences, which are extracted from executable files by using static analysis. As noted in Section 2, opcode sequences have been used in many research studies on malware detection. We use IDA Pro to disassemble files and extract the static opcode sequences from the result. For example, suppose we disassemble a portable executable file and obtain the ASM file. A snapshot from an ASM file is shown in Fig. 8.
Example disassembly [7].
The static opcode sequence corresponding to this disassembly is
Unigram: call, push, lea, push, push, call, add, test, jzBigram: call push, push lea, lea push, push push, push call, call add, add test, test jz
Ten samples of NGVCK family residues.
The following sequences, which are the beginning of some residues shown in Fig. 9, were generated from ASM files belonging to NGVCK malware family. Each base (symbol) in the residue can indicate different types of N-grams. The residues along with the consensus sequences (indicated by red) constitute the MSA matrix as can be seen in Fig. 10.
An MSA snapshot generated form NGVCK family.
We discard all operands such as labels and directives, and only retain the mnemonics (opcodes). For the top N frequent opcodes, N unique symbols were used and one symbol as a wild card for the rest. These unique symbols can be generated from single opcodes (unigram) or N general combinations of every N consecutive opcodes (N-gram). This is discussed further in Section 5.3. Then, sequences are used to train a PHMM, and the resulting model is used to score the files in the malware and benign families test sets. The scores from a given experiment are used to form a scatterplot.
In this section, we first discuss the tools that can be used to extract features from binary code. Then, we give an overview of the malware dataset used in this work. Finally, we elaborate on the experiments conducted. In Section 5.3, we provide the results of our experiments. All supplementary materials including the code, datasets, and results are available for public access on the Web.2
All supplementary materials including codes, datasets and full results are presented in
IDA Pro
This disassembler produces highly accurate assembly code from an executable file. It can also be used as a debugger. IDA is a powerful tool that supports scripting, function tracing, instruction tracing, instruction logging, and so forth. IDA Pro is used for static analysis, and, in particular, for the generation of “.asm” files from “.exe” files, from which opcodes and windows API calls can be extracted. Being a multi-processor and debugger, IDA Pro is available for various platforms such as Windows, Linux and Mac OS. Note that, there are Evaluation and Freeware versions of IDA freely available for non-commercial use with limited availability, but the Full version is not free of charge. We used the IDA Pro Disassembler version 6.8.1 for extracting opcode sequences from malware/benign family file sets.
PE explorer
Compared to other disassemblers, this tool has been designed to be more user-friendly. To achieve this aim, some of the components of other disassemblers have been removed which has resulted in an easy and rapid process. PE Explorer is the same as the other famous and proprietary tools for disassembling. In addition, it focuses on clarity, user-friendliness, and smoothness of navigation. PE Explorer is capable of opening, viewing and editing a variety of Windows executable file types (i.e. PE files). The evaluation version of PE Explorer is free of charge for 30 days with basic technical support.
Capstone
As said in Capstone webpage, “Capstone is a lightweight multi-platform, multi-architecture disassembly framework”. It comes with some exported methods helping to disassemble binary files. Some of its features are: Multi-architectures, providing details on the disassembler and some semantics of the disassembled instruction, and Native support for operating systems such as Windows, Mac, iOS, Android, and Linux. Capstone is free and available for well-known operating systems in package repositories. It is a Python/C library and using “pip install capstone” command, the Python module can easily be installed on Windows, Mac OS and Linux. The process of disassembling and debugging the portable executable files (for extracting features) and model generation and classification can be implemented as an integrated system using Capstone.
Datasets
Due to constant improvement and progress in development of new malware, a single benchmark cannot be used over a long period since using the models trained on these old datasets will not be desirable in malware detection field. Therefore, there is not one common dataset available as an assessment reference to all researchers. In this study, we used a collection of benign files from Cygwin related files. Additionally, we amassed a dataset containing four malware families used in several previous related work. The opcodes were extracted by the IDA Pro when not available. For each family, there are 50 examples of construction kits available on the VX Heaven website, with the exception of the NGVCK family of 200 samples, which is one of the most obscure malicious pieces of software available on malware detection because of their complex transformations. These datasets are also used in evaluations of various methods for malware detection that have been addressed in a number of previous works [1, 14, 31, 41, 51, 52, 53, 54, 55]. According to measurements of similarity carried out on the dataset by Rezaei et al. [14], after computing the similarity score using the method described in [41], samples in each family are compared in pairs. The least, the most and the average quantity of samples pair-wise comparison in each virus creation kit can be seen in Table 1.
Min, max, and average kits similarities percentage
Min, max, and average kits similarities percentage
NGVCK generates viruses with similarities ranging from 1.5 to 21.0% with an average of about 10.0%. This is a far lower degree of similarity than any of the other three generators. For the non-NGVCK generators, the similarity between two variants of the same virus ranges from 34.4 to 96.6%, and the average scores of G2, VCL32, and MPCGEN are 74.5, 60.6, and 62.7%, respectively. On the other hand, normal files give an average similarity of 34.7%. From these results, we can see that the NGVCK viruses are substantially different from one another while the virus variants generated by the other generators are more similar to one another than normal files. We conclude that the non-NGVCK generators we tested are not nearly as effective as NGVCK at generating metamorphic viruses.
To evaluate our models, we implemented a statistical-based malware detection technique based on profile hidden Markov models. In addition, we compared our results to n-gram analysis (with different
Attaluri et al. [1] proposed a method based on PHMMs. Since a different dataset was used by them, we implemented their method using Python and simulated the results on our dataset. The results of this method are provided in Fig. 11. As can be interpreted from the plot and also stated in the original paper, the method was not successful on classifying NGVCK family. In the evaluation phase, the Log Likelihood per Opcode (LLPO) is used to compute similarity score in order to detect a member of malware families (models). LLPO is computed by dividing the output of log likelihood function by the file’s length to create a length independent measure.
The simulated results of Attaluri’s proposed method using standard PHMMs (without consensus sequences).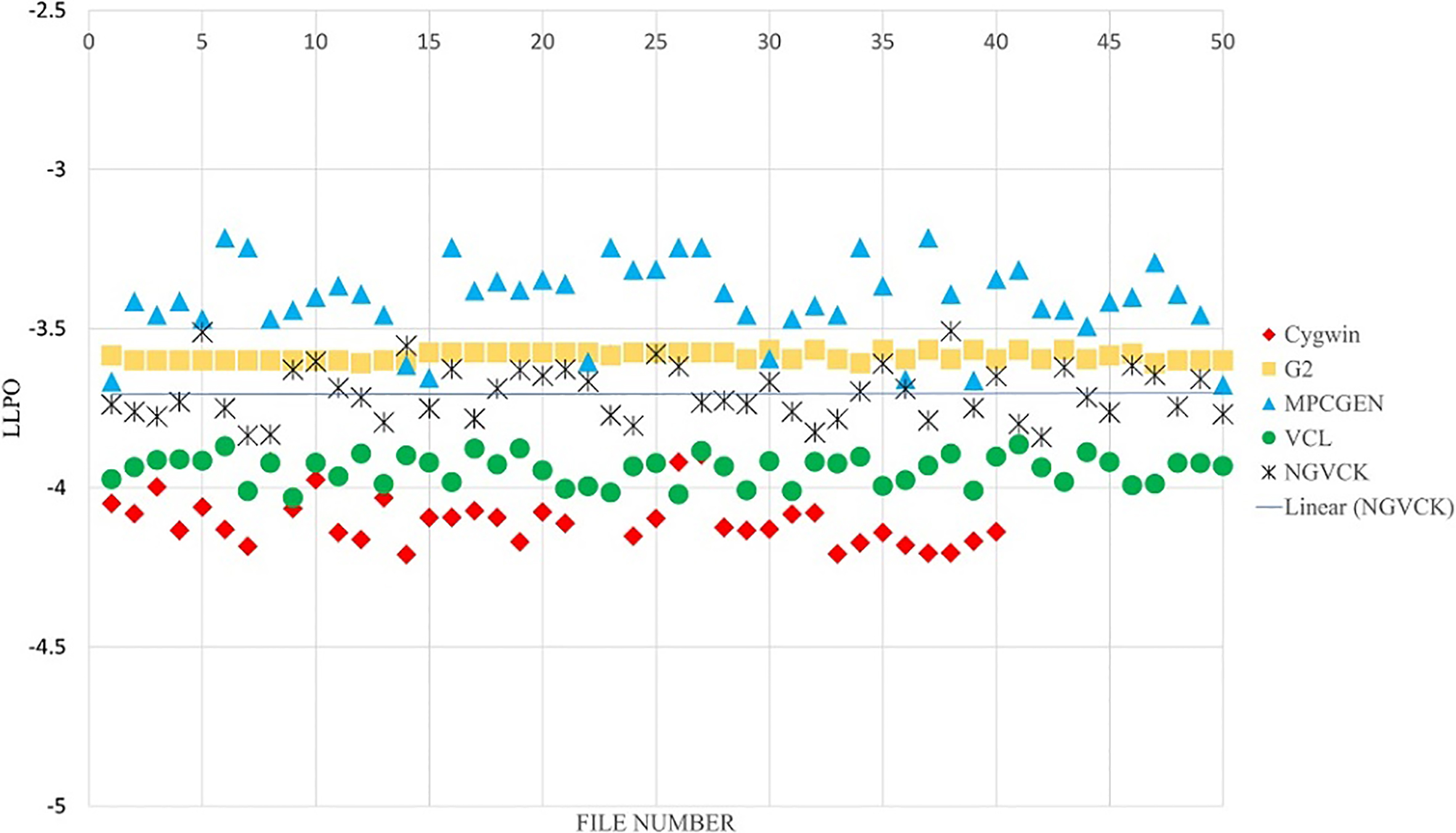
The strength of the proposed approach lies in adding consensus sequences to the MSA for building APHMMs. By adding the consensus sequence to the MSA, we build an advanced PHMM, which puts more emphasis on the frequent patterns existing in each family of malware. The effect of the number of consensus sequences is analyzed in the experiments reported in the various figures. Figures 12 and 13, present the result of classifying the NGVCK malware family with one and four consensus sequences, respectively. As mentioned before, for this family we had 200 sequences from which 150 sequences were used for training and 50 sequences were used for testing. The results for classifying other families are provided in supplementary material.3
The results of our proposed method for classifying NGVCK family by using APHMMs (one consensus sequence).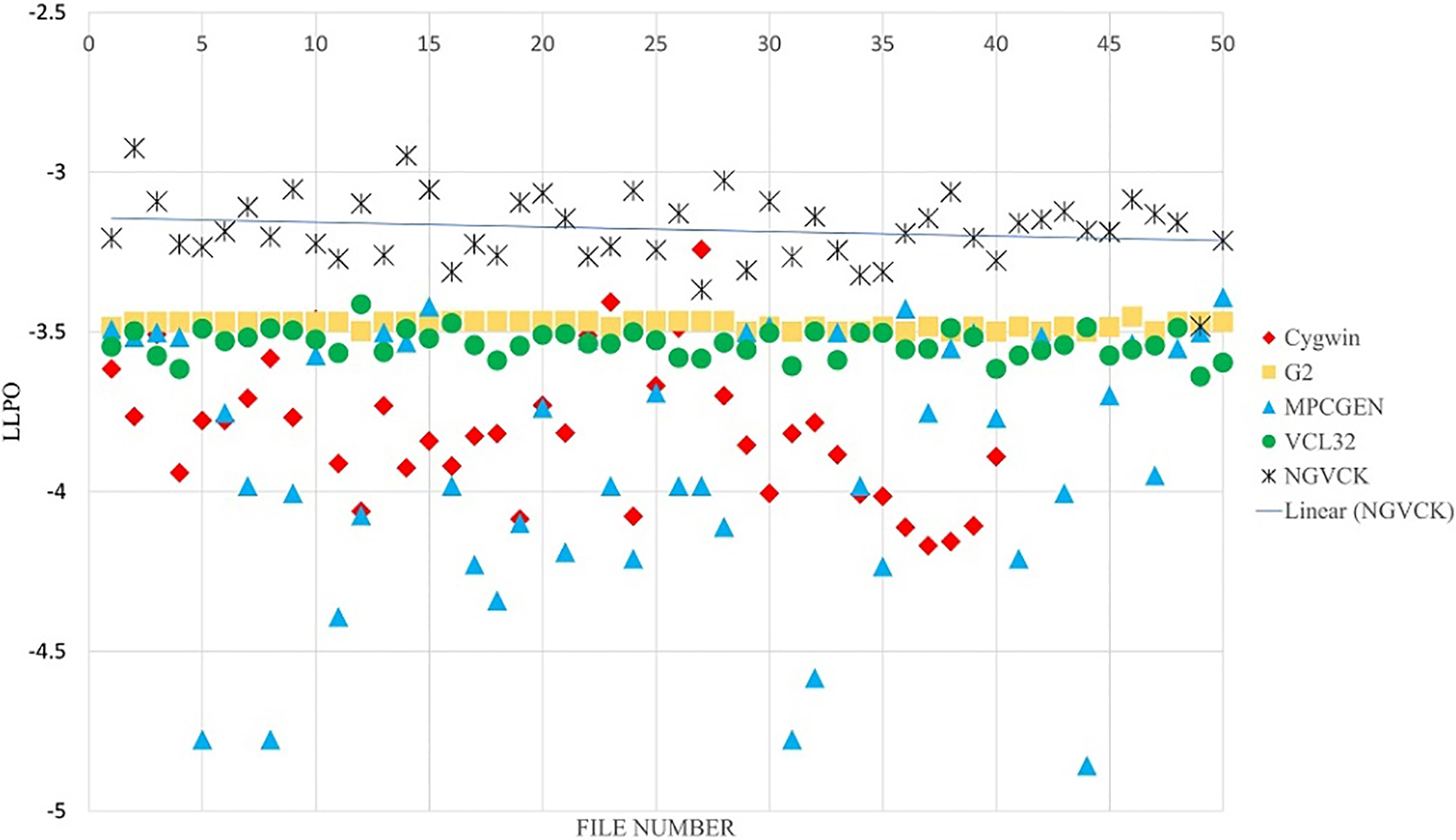
The results of our proposed method for classifying NGVCK family by using APHMMs (four consensus sequences).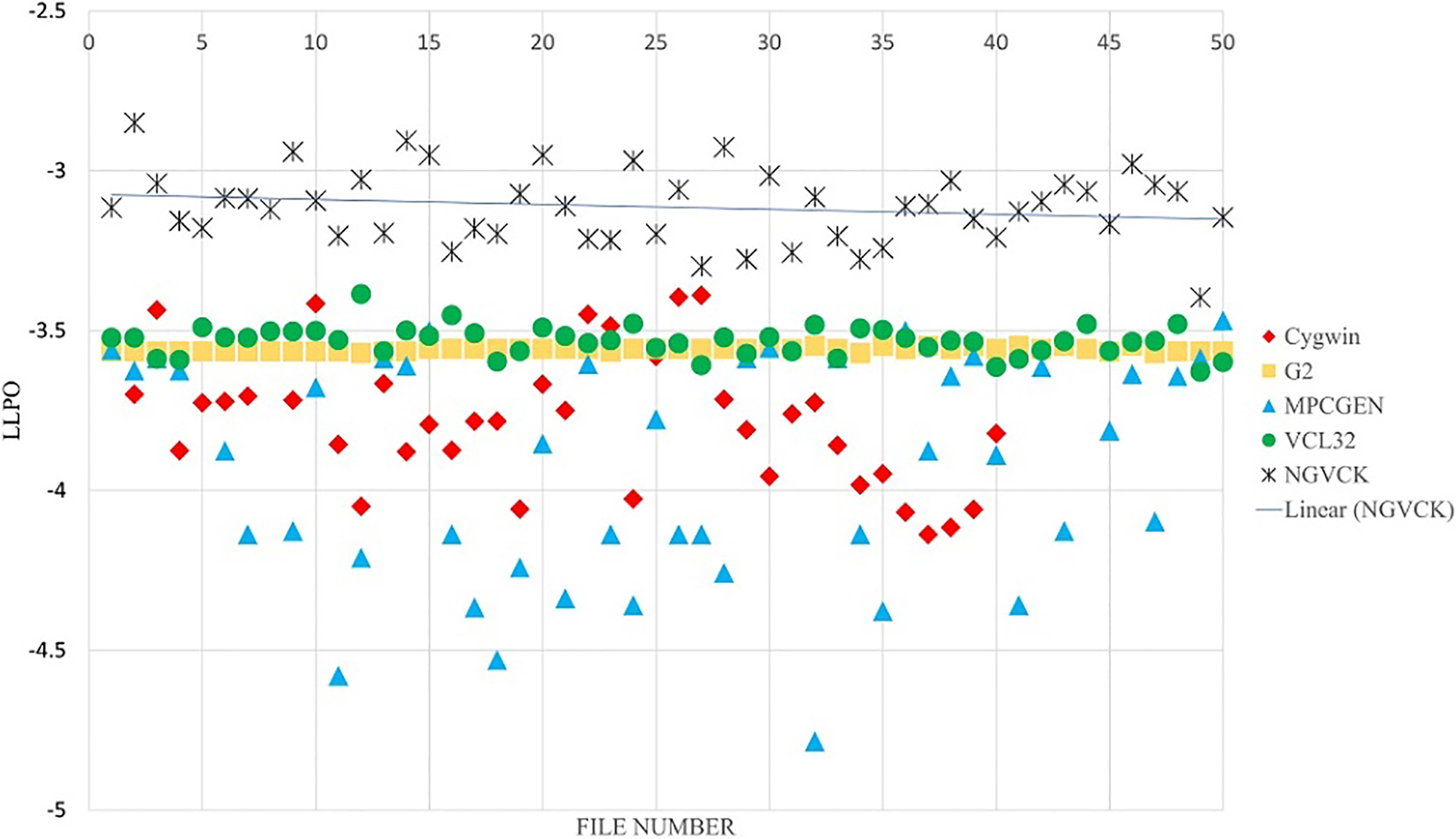
In feature extraction phase, we use a symbol for each frequent opcode. For the remaining opcodes, we use one symbol. In the former experiments, only top 36 unique opcodes are used (totally 37 opcodes). The results of unigram feature extraction method are presented in Figs 14 and 15 for 37 and 200 symbols, respectively. In Fig. 16, the results on bigram with 100 symbols are presented. By comparing Figs 14 and 15, we see that with unigram features using less symbols results in better distinction of the NGVCK class. If we analyze the frequency of all possible unigram symbols, we see that the top 14 opcodes cover 93% of all opcodes existing in all of the files with 98% of these opcodes covered by the top 50 opcodes [56]. We chose the top 36 opcodes as used in the research carried out by Attaluri et al. [1]. Having bigram symbols, the number of frequent ones will increase relatively. As we can see in Figs 14 and 15, having more symbols does not improve the results of unigram feature setting. The reason is the non-frequent symbols affects the scoring emission matrix and makes the score values unnecessarily small. For larger N values, the symbols are generated sequences from the combination of N successive opcodes, which are unique and informative (leading to better separation of families) even for very large numbers of symbols. Since PHMM is a position-specific model, using N-grams (N
The results of our proposed method for classifying NGVCK family by using one consensus sequence, unigram and 37 symbols.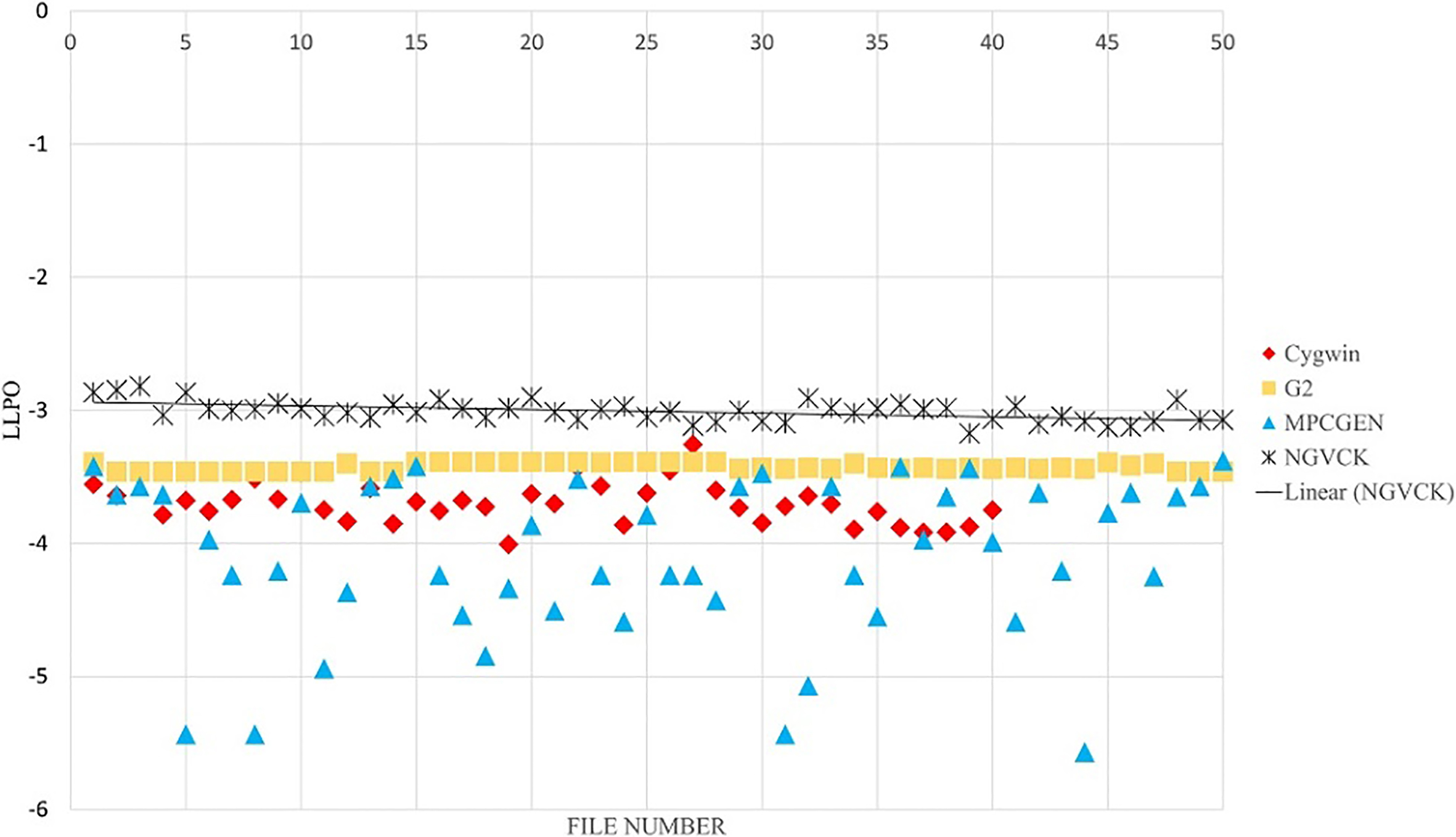
The results of our proposed method for classifying NGVCK family by using one consensus sequence, unigram and 200 symbols.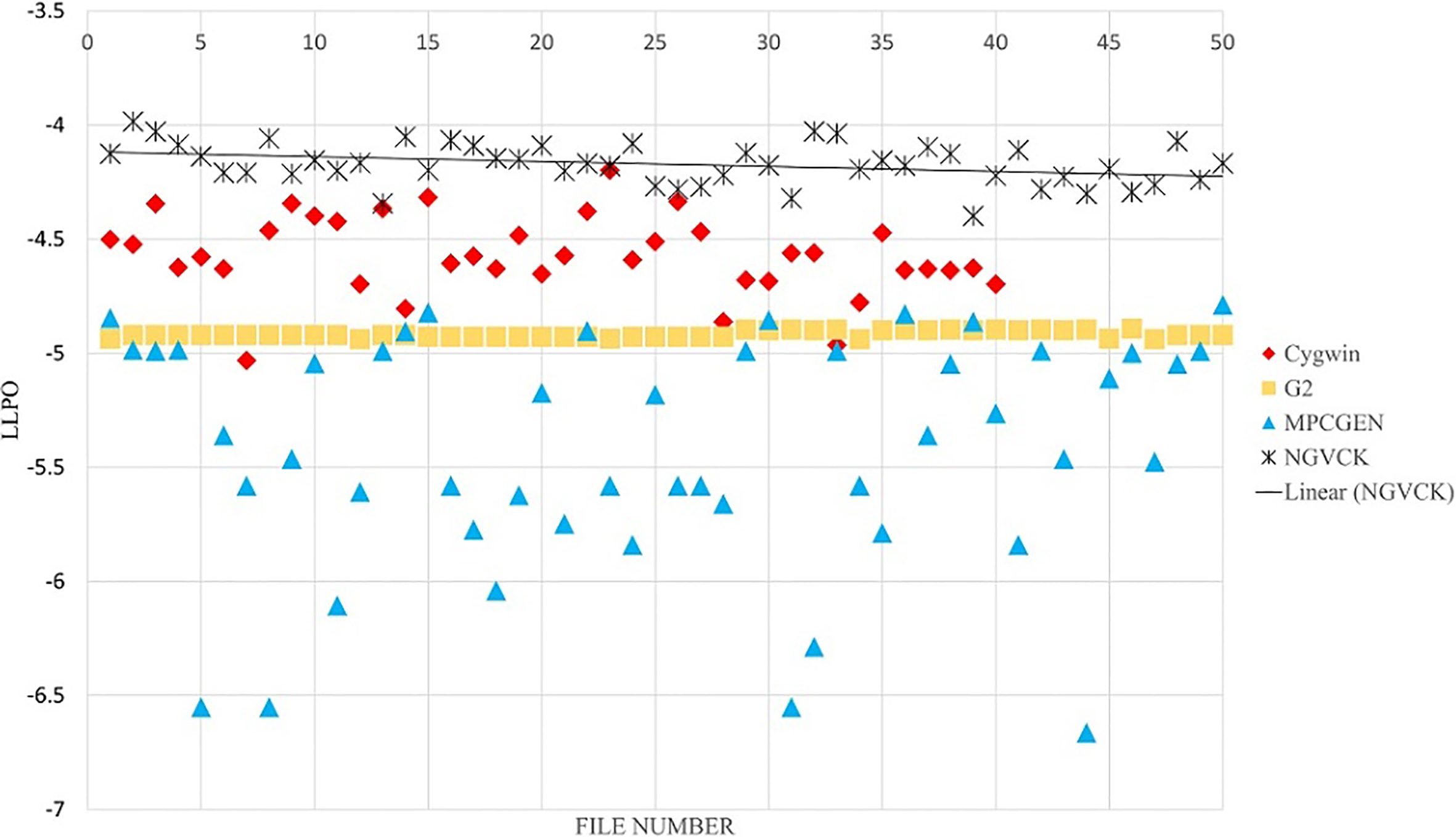
The results of our proposed method for classifying NGVCK family by using one consensus sequence, bigram and 100 symbols.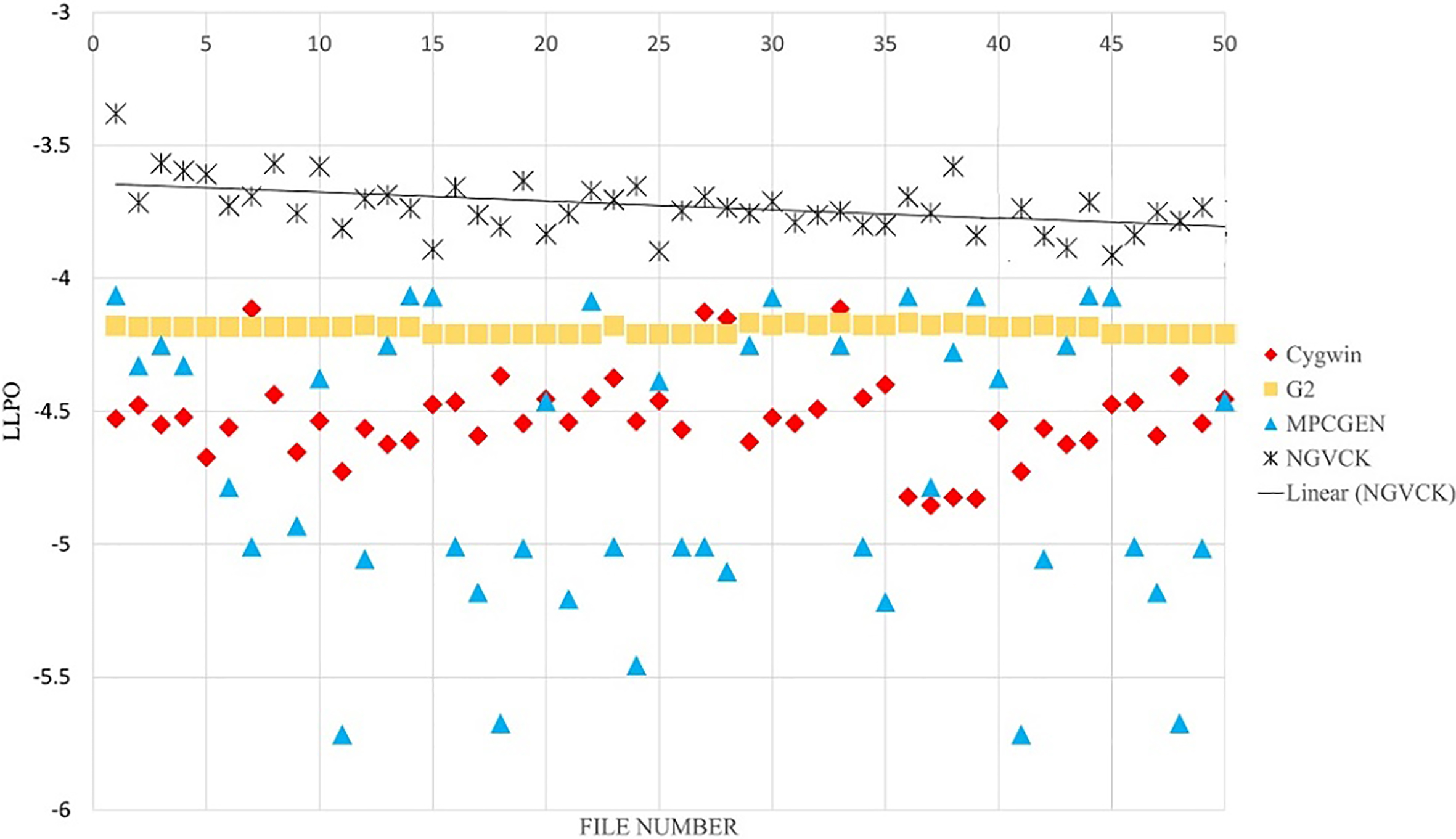
Table 2 shows a summary of all the results. We evaluate our results with three parameters, which directly impact on precision and recall. First is the number of consensus sequences (No. CSs) which plays an important role in the classification of malware families by emphasizing the common features. The second parameter (No. Syms) is the number of different symbols used in our MSA implementation and the third input parameter shows the number of consequence opcodes groups as an atomic part (unigram or bigram). The higher the order for this parameter, the better the classification. While higher orders of N-grams consist of more number of unique bases, it might make the classification better. Equations (1) and (2) show accuracy and FPR (False Positive Rate) respectively:
where, TP and TN mean True Positive and True Negative, respectively. FP stands for False Positive, and FN means False Negative.
The summary of our experimental results on NGVCK family where FPR means false positive rate, CSs stands for consensus sequence and Syms means symbols
We presented a separate experiment for each of the malware datasets listed in Section 5.2 for each of the various combinations of conditions mentioned in Table 2. For each experiment, we evaluated the effect of three parameters, the number of consensus sequences, the order of n-grams, and the number of symbols (bases).
Comparison of existing techniques with our method
In this paper, we designed and evaluated various malware detection techniques based on opcode sequences. We trained profile hidden Markov models and compared detection rates for models based on N-grams and consensus sequence approaches. Additionally, in order to obtain the perfect sensitivity score (i.e. one hundred percent), the false positive rate is 0.01 when using one consensus sequence in the model construction, and is reduced toward zero when four consensus sequences are considered. The evaluation results show that our classification technique obtains the perfect accuracy even when there are a few training datasets to build the models. We compare the performance of our advanced classification approach with some previous works. As one of our principal targets is high accuracy, we compare only with those methods which achieve the accuracy of 90% or higher on the set of executables. Table 3 summarizes this comparison.
Moreover, in the proposed approach presented in this study, we have leveraged reverse engineering and disassembly techniques in preprocessing and data preparation phases to extract the features needed to create our statistical model which increase the speed of the whole process. As a proposal for future research, another model could be considered to obtain the raw information from reading files in their binary form. By reading under question files byte-by-byte and in hexadecimal format instead of extracting information from opcodes, we may extract enough features to create the profile hidden Markov model. This process automatically minimizes the need for human intervention with the results and increases the processing speed especially in large-scale datasets analysis.
Footnotes
Acknowledgments
We would like to thank all the Artificial Intelligence Lab members at the Institute for Advanced Studies in Basic Sciences (IASBS) especially Dr. Zahra Narimani, Assistant Professor of Computer Science and Information Technology Department, IASBS, and Akbar Karimi, PhD student at the University of Parma, for their invaluable help in the process of developing this paper.