
Abstract
Research indicates that certain antihypertensive medications alter Alzheimer’s disease (AD) biomarkers in Caucasians. The renin angiotensin system (RAS) regulates blood pressure (BP) in the body and the brain and may directly influence AD biomarkers, including amyloid-β (Aβ) neuropathology, cerebral blood flow (CBF), and inflammatory markers. This hypothesis is supported by studies, including ours, showing that antihypertensives targeting the RAS reduce the risk and slow the progression of AD in Caucasians. While mounting evidence supports a protective role of RAS medications in Caucasians, this mechanism has not been explored in African Americans. To assess the mechanism by which RAS medications modify the brain RAS, cerebrospinal fluid (CSF) Aβ, CBF, and inflammatory markers in African Americans, we are conducting an eight month, Phase Ib randomized, placebo controlled trial, enrolling 60 middle-aged (45–70 years), non-demented individuals, at risk for AD by virtue of a parental history. Participants include normotensive and treated hypertensives that have never been exposed to a RAS medication. Participants are randomized (1 : 1:1) by gender and BP medication use (yes/no) to one of three groups: placebo, or 20 mg, or 40 mg telmisartan (Micardis), to determine the dose required to penetrate the CNS. Our overarching hypothesis is that, compared to placebo, both doses of telmisartan will penetrate the CNS and produce salutary, dose dependent effects on the brain RAS as well as CSF Aβ, CBF, and CSF inflammatory markers in African Americans, over eight months. This manuscript describes the trial rationale and design.
Keywords
INTRODUCTION
Certain antihypertensive medications appear to alter Alzheimer’s disease (AD) biomarkers in Caucasians and African Americans [1, 2]. The renin angiotensin system (RAS) regulates blood pressure (BP) and fluid maintenance in the body and the brain, and may directly influence AD biomarkers, including amyloid-β (Aβ) and tau, cerebral blood flow (CBF), and inflammatory markers [3–5]. This hypothesis is supported by studies showing that antihypertensives targeting the RAS, that most commonly are angiotensin converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (A2RBs), reduce the risk and slow the progression of clinical AD [6–8]. We have shown that 1) blood-brain barrier (BBB) crossing RAS medications exert protective cognitive effects and slow disease progression in hypertensive individuals with mild cognitive impairment (MCI), and 2) BBB crossing RAS medications are able to significantly alter brain RAS levels in middle aged Caucasians at risk for AD due to a parental history [9].
While mounting evidence supports a protective role of RAS medications in Caucasians, this mechanism has not been clinically tested in African Americans. We recently published a meta-analysis that reported the rate of AD incidence is 64% higher for African Americans than for Caucasians [10]. Moreover, we estimated the current U.S. AD prevalence for ages 65–90 to be 5.5% for Caucasians, and 8.6% for African Americans (prevalence ratio 1.56). Thus, not only are African Americans more likely to become afflicted with AD, they are also living longer with the disease. Because African Americans are at higher risk for AD than Caucasians, and RAS medications have shown to positively influence AD biomarkers and clinical disease progression, the potential benefit of RAS acting medications in this high-risk population needs to be addressed.
African Americans are at higher risk for vascular risk factors, including hypertension, which independently contributes to cognitive impairment and AD, possibly via RAS regulation [11]. African Americans have lower circulating renin and higher aldosterone and sodium levels than Caucasians, and therefore RAS antihypertensives, which are those medications believed to exert AD-related benefits in Caucasians, are not indicated for African Americans, though safety and efficacy studies of RAS acting medications show that they are well tolerated [12, 13]. If RAS medications are prescribed, higher doses are generally required to achieve comparable BP control to that seen in Caucasians at lower doses. Importantly, the dose needed to alter RAS levels in the brain is unknown, and may be lower than the dose required to alter BP, as suggested by observed benefits of an ARB on cognitive decline that were independent of changes in BP. While many researchers now argue that prescription practices based on race are unwarranted, these trends persist in clinical practice and were upheld in the JAMA Guidelines for the treatment of hypertension in 2014 [14]. Because we know that the brain RAS is implicated in AD neuropathology and we can manipulate this system in the brains of Caucasians [15], we are investigating whether RAS medications confer similar AD-related benefits in African Americans, and whether we can clinically alter this system, and at what dose.
We previously reported that individuals taking RAS acting medications (ACEIs or A2RBs), particularly those that are centrally acting, show slower cognitive decline and are less likely to progress from MCI to AD over three years [16]. However, the potential mechanism was unclear due to the absence of neuropathological findings, although results are consistent with studies of postmortem brain tissue of AD patients, that point to elevations of angiotensin converting enzyme (ACE), that both ACEIs and A2RBs are designed to mitigate [17]. More recently, we confirmed these results, and further reported that individuals taking RAS acting medications exhibited significantly fewer neurofibrillary tangles than non-RAS users in five brain regions including the mid-frontal cortex, mid-temporal cortex, inferior parietal cortex, entorhinal cortex, and hippocampal CA1 region (manuscript under review).
Importantly, because Aβ and tau accumulation and RAS dysfunction begin during midlife, it is imperative to investigate these biomarkers in middle-aged, presymptomatic adults, while there is time to intervene, before the irreversible AD cascade begins. Further, exploring the potential benefit of using existing drugs (i.e., “repurposing”) that are generally recognized as safe, such as RAS medications, shortens the time needed to demonstrate efficacy, and thus, more rapidly provides urgently needed treatmentoptions for this devastating illness.
Here we describe the rationale and design of the ‘Mechanistic Potential of Antihypertensives in Preclinical Alzheimer’s Trial’ (HEART). This trial is funded by the Alzheimer’s Association Program Part the Cloud Translational Research Funding for Alzheimer’s Disease Grant (PCTR) mechanism (PI: Wharton). The trial, which is coordinated at Emory University, began in March 2015. HEART is a Phase Ib double blind, randomized, placebo-controlled trial to determine whether RAS medications modify the brain RAS, cerebrospinal fluid (CSF) Aβ and tau, and CBF as well as central and peripheral inflammatory markers. This trial will recruit 66 middle-aged (45–65 years), non-demented African Americans who have a parental history of AD. Participants include normotensives and untreated and treated hypertensives. Participants are randomized by gender and antihypertensive use (y/n) to one of three groups: placebo, 20 mg telmisartan, or 40 mg telmisartan. Our overarching hypothesis is that, compared to placebo, both doses of telmisartan will penetrate the CNS andproduce salutary, dose dependent effects on the brain RAS as well as CSF Aβ and tau, CBF and CSF inflammatory markers, over 8 months.
STUDY DESIGN
Participants
Sixty-six non-Hispanic African American participants will be enrolled to achieve a final N = 60 assuming a 10% drop out rate over the 8-month trial. Both normotensive and hypertensive individuals are allowed to participate. Recruitment methods include the 1) Emory ADRC; 2) Emory Cognitive Neurology Clinics; 3) community based recruitment (churches, health fairs etc.); and 4) e-mail blasts to existing Emory research cohorts. Inclusion and exclusion criteria are outlined below. Parental history is confirmed by either autopsy-confirmation or probable AD as defined by NINDS-ADRDA criteria [18], if the participant’s parent was seen at an ADRC or the Emory Memory Clinic. Parental diagnosis is further verified using the validated Dementia Questionnaire and medical records, when available [19]. While subjective memory complaints, AD medications and Montreal Cognitive Assessment (MoCA) score are all used in an attempt to exclude individuals with MCI, lack of a clinical diagnosis and medical records for these participants is a limitation of the study.
Inclusion criteria
1) a biological parent with or who had AD; 2) African American; 3) ≥ 45 years of age; 4) willing to fast for 8 h; 5) willingness to undergo all procedures including blood draw, lumbar puncture (LP), and MRI; and 6) willingness to take a study medication daily for 8 months.
Exclusion criteria
1) Current or past (past 10 years) use of a RAS acting medication; 2) contraindication for LP or MRI; 3) significant neurologic disease; 4) heart failure; 5) type I or II diabetes; 6) history of significant head trauma; 7) major depression within the past two years; 8) HIV/AIDS; 9) history of alcohol or substance abuse; 10) significant systemic illness or unstable medical condition that could affect cognition or cause difficulty complying with or completing the protocol; 11) diagnosis of AD; 12), subjective cognitive impairment MCI or residence in a skilled nursing facility; 13) use of another investigational medication or AD medication; 14) unwillingness to fast.
Study medication rationale
The primary goal of this Phase Ib trial is to assess 1) the central activation and 2) the influence on AD biomarkers, by low and clinical starting doses of an FDA-approved RAS-acting medication. Participants taking a RAS medication currently or within the past 10 years (ACEI or ARB) are not eligible for enrollment. The clinical starting dose of telmisartan for BP control in Caucasians is 40 mg. This study will test the central activation of the drug at 20 mg versuss 40 mg versus placebo in African Americans, since they are commonly given higher RAS-acting medication doses to achieve BP control. Importantly, though African Americans are given higher doses for BP control, the dose required to cross the BBB and alter the brain RAS is unknown, and it is likely that this dose is lower than the dose required to alter BP in thebody.
Intervention and randomization
Standardized randomization by the study biostatistician is used to assign participants by gender and antihypertensive use (y/n) by the Emory Pharmaceutical Research Center to receive 20 mg telmisartan (n = 22), or 40 mg telmisartan (n = 22) or placebo (n = 22) to be taken once daily before bed. We chose to randomize by gender based on past studies enrolling African Americans, where we have an overrepresentation of women participants versus men. Yes/no was selected as the randomization classification for non RAS-acting medication use because the central question is RAS use versus placebo, thus we are not randomizing by medications that act via different mechanisms. Study drug and placebo are formulated into identical capsules. The PI, study personnel, and participants are blind to drug assignment. A study physician ensures BP control and is the only individual aware of drug assignment. The purpose of this Phase I RTC is not to control BP; therefore, we do not assume control of the participants’ BP maintenance as long as they are within safe limits. All participants remain on their medications as prescribed by their primary care physician (PCP) in addition to taking the study medication or placebo pill. All participants’ PCPs are sent a letter describing the HEART project at Baseline. Participants whose BP is too high or low at screening, or whose BP drops to unsafe levels during the study are referred to their PCP. In addition to the study medication (telmisartan or placebo), other non-RAS acting medications (e.g., HCTZ, calcium channel blockers, and beta blockers) are allowed. Once participants are enrolled, addition of RAS-acting antihypertensive medications is not allowed. Medication compliance is assessed using pill count at Month 4 and Month 8.
Trial description summary
Figure 1 shows the timeline and visit procedures of the HEART trial (NCT02471833). Details of each visit and procedures are also described below. Participants attend between 5–7 visits over the course of the 8-month trial depending on the number of safety visits needed. An initial phone interview determines eligibility and introduces the study protocol using standardized questions. Those who are eligible are invited for a screening visit, which takes approximately 1 h. If blood levels assessing kidney and liver function, blood cell count (potassium and creatinine), and BP values are within range (outlined below), individuals are asked to return for a Baseline visit and randomization. Prior to the Baseline visit, each participant’s primary care physician is mailed a letter outlining the HEART trial. Baseline visits are fasting, take approximately 6 h, and can take place on one day or split between 2 days. Participants begin taking the study drug after all Baseline procedures are complete. Participants are then seen after 2 weeks for a safety visit, which lasts approximately 30 min. If all blood levels and BP values are within range (see screen visit description below), participants return at Month 4. In the event that a blood measure or BP value is out of range, participants are advised to visit their PCP and asked to return for a second safety visit, and again referred to their PCP if out of range values persist and scheduled for a 3rd safety visit. If BP or blood measures remain out of range after the 3rd safety visit, the participant would not be permitted to continue with the trial. Participants return at Month 4 for drug refill, pill count, BP reads, and blood draw, which takes 1 h. At Month 8, participants return for their final fasting 6-h visit, which is identical to Baseline, except drug is not dispensed.
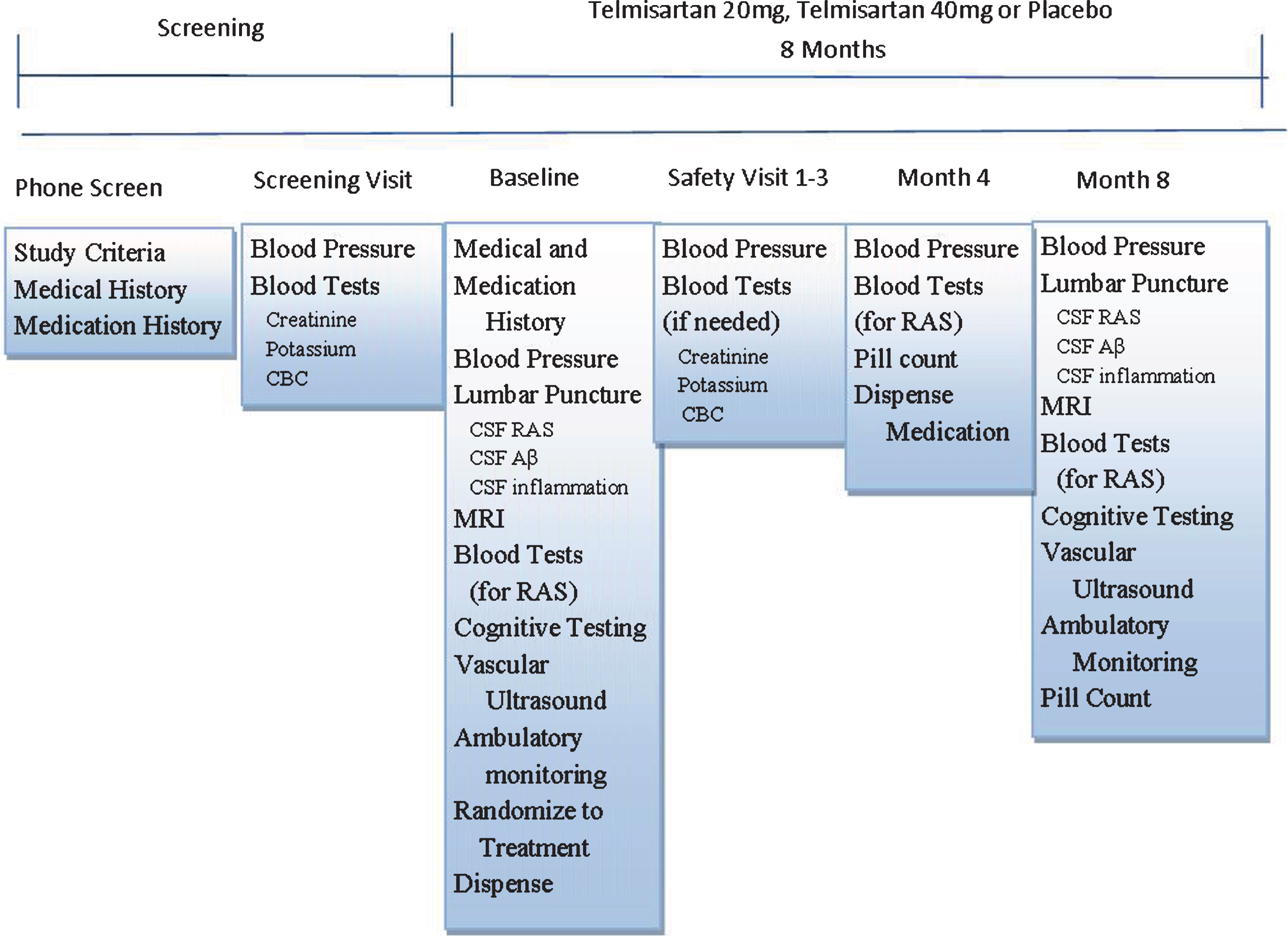
Timeline and visit procedures of the HEART trial. CBC, clinical blood count; CSF, cerebrospinal fluid; RAS, renin angiotensin system.
STUDY VISIT SPECIFICS
Screening
Initial eligibility is determined via phone interview by a member of the HEART team, at which time a screening questionnaire and the Dementia Questionnaire are administered [19]. This screening process explores a potential participant’s match against the inclusion and exclusion criteria described above, and eligible individuals are invited for a clinic-based face-to-face screening visit. The in-person screening visit includes obtaining informed consent, collecting medical and medication information, a cognitive screening questionnaire [20] two seated BP measurements, and a metabolic safety panel for hyperkalemia and renal insufficiency. Participants are unable to proceed to Baseline for the following: potassium >5.0 mg/dL or creatinine >1.99 mg/dL and/or systolic BP <110 mm/Hg or >170 mm/Hg, or MoCA score <26. Eligible participants are asked to return for randomization and a Baseline visit within 30 days.
Baseline and randomization
During this visit, BP, blood cell count, neuropsychological testing, physical exam, blood draw, MRI, LP, and vascular ultrasound are completed. Participants are randomized by PRC and begin study medication (or placebo) after this visit.
Safety (week 2) visits
Following Baseline, participants are seen at Week 2 for BP assessment, metabolic blood panel, heart rate, symptom checklist, adverse events questionnaire (AE), and health history and medication history update. While potassium and creatinine cut-off criteria are the same as Screening, systolic BP at Safety and Month 4 visits must be ≥ 90 mm/Hg. If all measures are within range, the participant returns at Month 4. If BP or blood levels are not within limits, the participant is advised to schedule a visit with their PCP for treatment. The participants are then asked to return in 2 weeks for another safety visit. If a participant’s BP or blood levels do not improve by three Safety Visits, they would still be able to continue in the trial, but they are not allowed to continue on study drug.
Month 4 visit
This midpoint visit lasts 1 h and includes pill count and medication dispensing, AE questionnaire and symptom checklist, blood draw, and BP measures.
Month 8/study completion
The final 6 h visit includes: BP, AE questionnaire, symptom checklist, pill count, neuropsychological testing, neuroimaging, vascular ultrasound, LP, and blood collection. This is a fasting visit and is identical to the Baseline visit except for medication dispensing.
PROCEDURE DESCRIPTIONS
Arterial function assessment via ultrasound: PWV
Pulse wave velocity (PWV) is non-invasive and is administered at Baseline and Month 8. This is a measure of arterial stiffness, which has been shown to be superior to clinical BP measures when predicting cognitive decline in healthy populations and may be more sensitive to BP medication changes than other measures [21]. PWV is a highly reproducible measure of artery wall stiffness and is strongly correlated with cardiovascular events and all-cause mortality. Both PWV and PWA are assessed using measurements of the radial, carotid, and femoral arteries, using the SphygmoCor Pulse Wave Velocity system (PWV Medical). In brief, peripheral pressure waveforms were recorded from the radial artery at the wrist, using applanation tonometry with a high-fidelity micromanometer. After 20 sequential waveforms are acquired, a validated generalized transfer function is used to generate the corresponding central aortic pressure waveform, and the degree of pressure augmentation due to the reflected wave form from the periphery is measured. Central AIx is calculated as the augmented pressure divided by the central pulse pressure from the central pressure waveform. Carotid-femoral artery PWV is determined by measuring transcutaneous Doppler flow velocity recordings carried out over the common carotid artery and the femoral artery.
Ambulatory blood pressure monitoring
Ambulatory BP is assessed at Baseline and Month 8 and endpoints include mean 24 h ambulatory BP and nocturnal dipping percentage defined as:
Cerebrospinal fluid collection
A after an 8-h overnight fast, CSF is acquired via LP the morning of the Baseline and Month 8 visits. CSF collection is completed according to guidelines put forth in the “Biospecimens Best Practice Guidelines for the ADCs” published by the National Alzheimer’s Coordinating Center (NACC). Participants are placed in the sitting position and asked to maximally flex their knees, hips, back, and neck. The skin over L4-L5 is prepped and draped in a sterile manner. 1% lidocaine is used as a local anesthetic, followed by insertion of a spinal needle with introducer into the L4-L5 interspace using sterile technique. Using a 24-gauge Sprotte needle and a gentle extraction technique, approximately 22 ml of CSF is collected using sterile polypropylene collection tubes. Samples undergo a 10-min 2500 rpm spin and are aliquoted into 500μl polypropylene cryovials and stored at –80°C for future assays, which will take place after all participants complete Baseline LP and bathed and tested again after all Month 8 CSF is collected.
CSF biomarkers
RAS measures: To determine the extent to which telmisartan may penetrate the CNS, we will analyze key RAS components (such as ACE, angiotensin I, II, III, IV, and ACE2) at Baseline and Month 8. We will use established methods including ELISA and activity assays using specific fluorogenic substrates [23–25] which, when coupled with the RAS measures collected in plasma, will provide the most comprehensive characterization of systemic and brain RAS function in African Americans to date. Tau and Aβ: CSF T-tau concentration is determined using a sandwich ELISA (Innotest hTAU-Ag, Innogenetics, Ghent, Belgium) specifically constructed to measure all tau isoforms irrespective of phosphorylation status [23]. Tau phosphorylated at threonine 181 (P-tau) was measured using a sandwich ELISA method (INNOTEST® PHOSPHO-TAU (181P), Innogenetics, Ghent, Belgium) [24]. Aβ1–42 levels will be determined using a sandwich ELISA (INNOTEST® β-AMYLOID(1–42), Innogenetics, Gent, Belgium), specifically constructed to measure Aβ containing both the first and 42nd amino acid [25]. Samples will be assayed in one batch after study completion by Dr. Zetterberg, an expert in CSF AD biomarkers. Dr. Zetterberg has performed all of our CSF biomarkers in this same ‘at risk’ cohort. All assays are performed as singlicates because the measurements are well standardized. Assay stability is monitored throughout the sample series using quality control samples representing low and high biomarker concentrations. Plates with CVs of the quality samples higher that 15% will be re-analyzed. All assays are conducted by board-certified laboratory technicians blinded to participant clinical characteristics and randomizationgroup. Immunophenotyping and inflammatory markers: These data will be used to test the hypothesis that RAS function is linked to peripheral and central inflammation as well as immune cell profile regulation. Inflammatory cell phenotyping will be performed via flow cytometry. We have successfully collected sufficient amounts of cells from CSF for analysis of immune cell subsets such as monocytes and other leukocytes. Assessment of cell-type specific markers allow quantification of monocyte, B-cell, and T-cell (CD4+ and CD8+) frequencies at Baseline. Additional markers of immune cell activation are obtained ex vivo in plated cells following incubation with various activating ligands and will allow sensitive detection of immune dysregulation. This novel method for CSF immunophenotyping provides invaluable information regarding the changes in immune cell frequency and their activation responses that occur in individuals at high risk for AD [26].
Neuroimaging
Participants undergo a 1-h MRI at Baseline and Month 8. Scan protocol includes structural MRI (e.g., T1-weighted imaging, Diffusion Tensor Imaging, T2, and T2FLAIR) and blood flow based measures (e.g., ASL perfusion, resting BOLD, and activation BOLD). MRI data will be acquired using a Siemens Tim Trio 3.0-Tesla scanner (Siemens Inc., Erlangen, Germany). The protocol included a 3D T1-weighted magnetization-prepared rapid acquisition with gradient echo (MPRAGE) anatomical imaging (TR/TI/TE = 2300/900/3.0 ms, FA = 9°, voxel size = 1×1×1.2 mm3, 176 slices). Subcortical volumes were extracted using a user-independent parcellation process in Freesurfer (version 5.03, MGH, Charlestown, MA) running on a Centos 6.6 operating system. Technical details of this protocol have been described previously in detail [27]. As an overview, nonbrain tissues are removed and a Tailairach transform is applied. Subcortical white matter and deep gray matter structures are parcellated by combining data from voxel intensity, probabilistic atlas locations, and local relationships between anatomical structures. This information is used to automatically assign each voxel to a neuroanatomical label. A Bayesian model based on prior manual tracings is applied. Lesion segmentation tool (LST) will be used to segment and quantify volume of white matter hyperintensity.
Regions of interest
Several of our analyses focus on specific brain regions of interest (ROIs) including regions that our preliminary studies indicate are affected preclinically. These include: 1) posterior cingulum bundle (PCB), 2) the portion of the cingulum bundle that runs between PCB and hippocampus, 3) retrosplenial white matter, 4) entorhinal white matter, 5) white matter adjacent to hippocampus, 6) fornix, and 7) splenium of the corpus callosum. Broader white matter analyses use voxel-wise methods, allowing us to test for effects in long (and late-myelinating) fiber bundles such the superior longitudinal fasciculus, which are known to be affected in AD.
Blood collection
Participants undergo blood draw for 1) RAS function assessment including aldosterone, angiotensinogen, renin and ACE activity and levels; 2) APOE genotyping; 3) inflammatory markers in plasma (MSD V-PLEX Pro-inflammatory Panel 1); 4) peripheral blood mononuclear cell (PBMC) immunophenotype. RAS function: We measure, using established methods [23, 25], RAS components that focus around the generation (ACE) and degradation (ACE2) of angiotensin II to determine the levels of RAS activity that is mediated through angiotensin II, the primary mediator of RAS. ApoE genotyping: Venous blood is collected into EDTA anticoagulated tubes and genomic DNA is isolated by standard protocols. We isolate 50 to 70 g of DNA from 2 mL of whole blood. APOE genotypes are determined by real-time polymerase chain reaction using TaqMan probes (Applied Biosystems Inc.) unique for each APOE single-nucleotide polymorphism, rs429358 (assay ID C3084793 20) and rs7412 (assay ID C 904973 10), according to established protocols. Inflammation and immunophenotyping: Using flow cytometry markers designed for the Human Immunophenotype Project (Stanford University), alterations of peripheral immune cell profiles in B cells (CD19), T cells (CD3, CD4, CD8), dendritic cells (CD11c, CD123), monocytes (CD14, CD16) and their activation status is assessed. Inducibility of MHC-II (HLA-DR and HLA- DQ) in monocytes treated with IFNγ is also assessed along with cytokine secretion in the conditioned media (MSD human V-PLEX Pro- inflammatory panel). We have previously reported an association between inflammation and cognitive performance in African Americans than Caucasians, and have recently reported higher levels of inflammation in both blood and CSF in a middle age cohort at risk for AD [26, 28].
Neuropsychological testing
Testing is being conducted to determine the potential impact of telmisartan versus placebo on cognition over eight months. Neuropsychological testing lasts approximately 1 h and includes both paper and pencil and computerized tasks. Alternative testing forms are not available for cognitive tests included in the present study, so the potential for practice effects may exist. Of all the cognitive domains, executive function is particularly vulnerable to vascular dysfunction and is impaired in early AD associated cognitive decline [29, 30]. Executive function is assessed using the NINDS-initiated EXecutive Abilities: Measures and Instruments for Neurobehavioral Evaluation and Research or “EXAMINER” tool box. This test battery reliably and validly assesses executive function in clinical trials (https://memory.ucsf.edu/examiner) [31–33]. Additional tests include measures of overall cognitive status (MoCA; which is also given at Screening) [20], memory (Benson Figure [34], Buschke Selective Reminding Test [35]), attention/set shifting (Trails A & B [36], Digit Span) [37], visuospatial functioning (Mental Rotation Test) [38], and language (Multilingual Naming Test) [39].
Questionnaires
The following information is collected from participants at Baseline and Month 8. Most questionnaires are computerized using the RedCap system and emailed to participants before their Baseline and Month 8 visits, so participants can complete them at their leisure and to cut down on study visit duration. Questionnaires include information pertaining to: 1) medical and medication history; 2) exercise [40]; 3) sleep [41]; 4) nutrition [42]; 5) positive [43, 44] and negative (stress) aspects of caregiving [45–47]; 6) depression [48]; and 7) behavior and quality of life of their parent with AD [49, 50].
Adverse events and data safety and monitoring
HEART is approved by the Emory University Institutional Review Board. Participants are screened for AEs via self-report at all visits. Questions and blood work regarding potential medication related symptoms include dizziness, light-headedness, weakness, lower extremity edema, hypotension, renal insufficiency, and hyperkalemia. Safety procedures are implemented in accordance with NIH safety policies for clinical trials. The safety monitoring is conducted by the PI the study physician, and a Data and Safety Monitoring Board (DSMB). The DSMB is comprised of three members including two physicians familiar with RCTs and African American hypertension studies and an experienced biostatistician.
Analyses and power calculations
We will conduct Wilcoxon-Mann-Whitney tests to assess whether the change in the level of each CSF biomarker (Month 8 versus Baseline) differs between the treated group and placebo group. Tests will be conducted separately for each dose group (20 mg and 40 mg) but we will also consider testing for a dose-response by conducting Jonckheere-Terpstra trend tests. P-values <0.05 will be regarded as significant for the primary outcomes (CSF Aβ, tau, ACE). We will use False discovery Rate (FDR) correction to adjust p values for all secondary outcomes. We will use a 0.025 alpha level for all outcomes, which is in accordance with the FDR. We will consider linear regression analysis (outcome is 8-month change in a biomarker) to adjust for demographic or clinical confounders. Demographic covariates will include age, education, and gender. Clinical covariates will correspond to the appropriate outcomes, and will include, but are not limited to, ApoE ɛ4 status, BP, and depression. Power calculations for ACE, CBF, Aβ, and tau comparisons are based on data from previous trials in similar populations. With 20 participants per group, we will have 99% power to detect change in ACE levels and 80% power to detect differences in CSF Aβ, tau and CBF between the treated groups and placebo (based on 2-sided tests and Type I error rate α= 0.05).
CONCLUSION
With the increasing incidence and prevalence of AD, research efforts should target high-risk groups in order to prevent or slow disease progression in tandem with cure-driven research directives in established disease. Repurposing existing medications that have been shown to be beneficial in observational and clinical trials is a cost effective way of slowing AD, particularly in high-risk individuals, and allow for more rapid research trajectories over conventional drug discovery approaches [51]. While longer interventions are optimal in healthy controls, early intervention is critical and assessing the impact on biomarkers in addition to cognitive changes is also of great importance. Because we know that the brain RAS is implicated in AD neuropathology in Caucasians, research should clinically investigate the extent to which RAS medications confer AD-related benefits in African Americans via the same mechanism, and the rate at which they do so. The HEART trial will provide data to address these issues.
Footnotes
ACKNOWLEDGMENTS
The National Institute on Aging (K01AG042498 and 1RF1AG051514-01) and the Emory Alzheimer’s Disease Research Center (NIH-NIA 5 P50 AG025688) supports this project. We thank the HEART research participants for their willingness to devote their time to research, and the staff members who work tirelessly to make the research possible.