
Abstract
Defects in motor protein-mediated neuronal transport mechanisms have been implicated in a number of neurodegenerative disorders but remain relatively little studied in Alzheimer’s disease (AD). Our aim in the present study was to assess the expression of the anterograde kinesin superfamily motor proteins KIF5A, KIF1B, and KIF21B, and to examine their relationship to levels of hyperphosphorylated tau, amyloid-β protein precursor (AβPP), and amyloid-β (Aβ) in human brain tissue. We used a combination of qPCR, immunoblotting, and ELISA to perform these analyses in midfrontal cortex from 49 AD and 46 control brains. Expression of KIF5A, KIF1B, and KIF21B at gene and protein level was significantly increased in AD. KIF5A protein expression correlated inversely with the levels of AβPP and soluble Aβ in AD brains. Upregulation of KIFs may be an adaptive response to impaired axonal transport in AD.
INTRODUCTION
Neuronal health in the central nervous system is dependent on a range of physiological processes which maintain cellular structure, ion homeostasis, electrical activity, and synaptic function. The transport of cellular organelles and proteins throughout the neuron, including along the axon and dendrites, is crucial for maintenance of neuronal structure and function. Defects in motor protein-mediated neuronal transport mechanisms have been implicated in a number of neurodegenerative disorders [1].
Transport within neurons is mediated by multiple motor proteins, many of which are involved in synaptic transmission and axonal trafficking. Kinesin superfamily proteins (KIFs) comprise a large group of motor proteins whose primary role is the anterograde axonal transport and intraneuronal transport of protein cargoes through association with microtubule ‘rails’ [2]. Currently, there are 45 known members of the KIF family, 38 of which are neuronally enriched [3]. The majority of KIFs have an NH2-terminal head, comprising a microtubule binding domain and a conserved globular motor domain which hydrolyses ATP to produce the energy required for movement of cargoes along the microtubule and through the axon. The head is attached by an α-helical stalk to a COOH-terminal (tail) to which cargoes bind [2, 4]. In most cases, the tail domain of each KIF determines its cargo specificity. In terms of conventional kinesin, cargoes can bind directly or indirectly, through kinesin light chain (KLC) associations and protein adaptor complexes, to the tail domain [5]. Essential neuronal cargoes transported by KIFs include cellular organelles such as mitochondria, pre- and post- synaptic membrane proteins, and a range of structural proteins such as neurofilaments (NFs) [1].
Given the essential role of KIFs in protein trafficking, it is not surprising that mutations in KIFs are associated with neurodegenerative diseases. Point mutations in the KIF5A gene have been linked to several axonopathies including hereditary spastic paraplegia type 10 (SPG10) [6] and Charcot-Marie Tooth disease type 2A (CMT-2A) [7]. SPG10 is implicated in disturbed intracellular axonal transport and is characterized by axonal loss in the corticospinal tract [6, 8]. Animal gene knock-out studies have also highlighted the importance of KIF5A in axonal transport and neuronal development [9]. In addition, single nucleotide polymorphisms (SNPs) within the KIF5A gene locus (rs12368653 and rs703842) have also been linked to multiple sclerosis (MS) susceptibility [10, 11], and we previously found levels of KIF5A protein in MS tissue to be related to SNP copy number [12, 13]. KIF5A is believed to transport amyloid-β protein precursor (AβPP), mitochondria, and a range of pre-synaptic membrane proteins that form the SNARE complex [9, 14–18 , 9, 14–18]. The majority of these cargoes bind indirectly to KIF5A through KLCs.
SNPs in the region of the KLC1 gene have been associated with Alzheimer’s disease (AD) susceptibility [19]. Andersson et al. [20] reported that SNPs in KLC1 were associated with APOE ɛ4 carrier status and with the cerebrospinal fluid level of hyperphosphorylated tau in mild cognitive impairment patients who converted to AD during follow-up; suggesting that variability in axonal transport could influence early AD pathogenesis. In support of this theory, studies using familial AD (FAD) AβPP overexpressing transgenic mice have shown abnormal axonal morphology and large axonal swellings co-positive for KLC and phosphorylated NF-H within 4 months of birth [21]. In addition, transgenic mice with modified presenilin-1 expression have demonstrated reduced affinity between KLC and membrane bound organelles (such as synaptophysin and syntaxin-I containing vesicles), mediated via KLC phosphorylation by elevated glycogen synthase kinase 3β (GSK-3β) activity. Elevated GSK-3β activity has also been directly associated with levels of hyperphosphorylated tau in FAD models [22, 23]. The formation of intraneuronal neurofibrillary tangles (NFTs), composed of aggregated hyperphosphorylated tau protein, is a cardinal feature of AD. Under physiological conditions tau interacts with tubulin as a microtubule stabilizer [24], forming an important contributor to the structure and integrity of the axon for signal conductance. We have previously found reduced KIF5A, KIF1B, and KIF21B expression in multiple sclerosis tissue [12] and demonstrated significant inverse correlations between KIF5A and cargo expression, suggesting that lower levels of KIF5A contribute to the axonal aggregation of proteins that lead to the formation of axonal spheroids, commonly seen in the disease [13]. KIF1B and KIF21B were studied as they have been linked to MS susceptibility [25, 26]. KIF1B is believed to share functional redundancy with KIF5A for neuronal protein cargoes and a mutation in its motor domain is also linked to CMT-2A [27]. KIF21B is dendritically enriched and involved in post-synaptic protein transport [28].
As intraneuronal aggregation of proteins is also a feature of AD, a disease in which neuronal protein transport is likely impaired [29], we thought it would be of interest to assess KIF5A, KIF1B, and KIF21B expression in AD and to analyze the relationship between these KIF motors and the levels of AβPP, Aβ, and hyperphosphorylated tau.
MATERIALS AND METHODS
Study cohort
Samples of midfrontal cortex (BA 9) cDNA and protein homogenate were obtained from the South West Dementia Brain Bank (Bristol, UK), under the terms of South West –Central Bristol Research Ethics Committee approval no 08/H0106/28+5. We studied 49 cases of AD (in which, according to the National Institute on Aging –Alzheimer’s Association criteria [30], AD neuropathological change was an adequate explanation of dementia) and 46 age-matched controls without a history of cognitive impairment (Table 1).
Clinical characteristics of Alzheimer’s disease and control patient cohort
Cases highlighted in Bold (postmortem delay >72 h) were removed from statistical analysis. AD, Alzheimer’s disease; C, control; F, female; h, hours; M, male; y, years.
Quantitative real-time PCR (qPCR)
Quantitative real-time PCR was performed on a StepOnePlustrademark Real-Time PCR system with StepOne software v2.1 (Applied Biosystems; Fisher Scientific UK Ltd, Loughborough, UK). Control and AD cDNA samples were used at a concentration of 2 ng/μL, diluted to a final volume of 20μL with TaqMan® 2x Fast Advanced Master Mix and TaqMan® gene expression assays for KIF5A (Hs01007893_m1), KIF1B (Hs01114538_m1) and KIF21B (Hs01118428_m1; FAM-MGB dye-labelled) (Applied Biosystems). qPCR was performed on a FAST ramp speed holding at 50°C for 2 min, 95°C for 20 s, followed by 40 cycles at 95°C for 1 s and 60°C for 20 s. Gene expression (2–ΔΔct) was calculated relative to the gene encoding the neuron-specific protein NeuN (RBFOX3; Hs01370653_m1; FAM-MGB dye-labelled) and neuron-specific enolase 2 (ENO-2; Hs00157360_m1; FAM-MGB dye-labelled) (AppliedBiosystems).
Western blotting
All primary antibodies that we used in dot blots were initially tested for antibody specificity by western blot. Protein homogenates were diluted 1 : 1 with Laemmli 2x sample buffer (Sigma-Aldrich Ltd; Dorset, UK) and heated to 95°C for 5 min to denature the protein before applying to gels. A Mini-PROTEAN Tetra Cell was constructed with mini-Protean TGX gels (4–20%) (Biorad Hertfordshire, UK). The Tetra Cell chambers were filled with Tris/Glycine/SDS running buffer (Biorad), before we loaded 7μL BLUeye Prestained Protein Ladder (Geneflow; Staffordshire, UK) and 15μL of denatured AD protein homogenate in the remaining lanes. The gel was run for approximately 1 h (until the dye front reached the gel bottom) at 150 V. Proteins on the gel were then transferred onto nitrocellulose membrane for 90 min at 350 mA. Nitrocellulose membrane from the gel transfer was blocked in 5% BSA/Tris-buffered saline-Tween 20 (TBS-T) or 5% milk/TBS-T (depending on antibody), for 1 h at room temperature before its incubation with primary antibody (reconstituted in membrane blocking solution), overnight at 4°C. Primary antibodies used for blotting were as follows: rabbit anti-KIF5A (Sigma-Aldrich Ltd; HPA004469), mouse anti-GAPDH (Abcam; Ab9484); mouse anti-AβPP (Zymed; Life Technologies Ltd; Paisley, UK; 13–0200), rabbit anti-KIF21B (Sigma-Aldrich Ltd; HPA027274), rabbit anti-NEUN (Abcam; Ab177487), mouse anti-PHF-TAU (Invitrogen; Fisher Scientifc UK Ltd; MN1020), and rabbit anti-KIF1B (Bethyl Laboratories; Montgomery, USA; A301-055A). Optimal antibody concentration and the specific blocking solution used are detailed in Table 2. Bound primary antibody was detected by incubation with HRP-conjugated goat anti-rabbit IgG pre-adsorbed (1 : 10,000; Ab6721) or goat anti-mouse IgG (1 : 5000; Ab6789) secondary antibodies (both Abcam), for 1 h at room temperature. Protein expression was visualized using a chemiluminescence EZ-ECL kit (1 : 1; Geneflow), in conjunction with a Biorad Universal III Bioplex imager. Densitometric band analysis was performed using Image Labtrademark 5.0 software (Biorad). All antibodies displayed specific bands as described on manufacturer data sheets and consistent with their reported molecular weights (Supplementary Figure 1).
Antibodies used for immunoblotting
AβPP, amyloid-β protein precursor; BSA, bovine serum albumin; DB, dot-blotting; KIF, kinesin superfamily protein; NeuN, neuronal nuclei; PHF, paired helical filament; TBS, Tris-buffered saline; WB, western blotting.
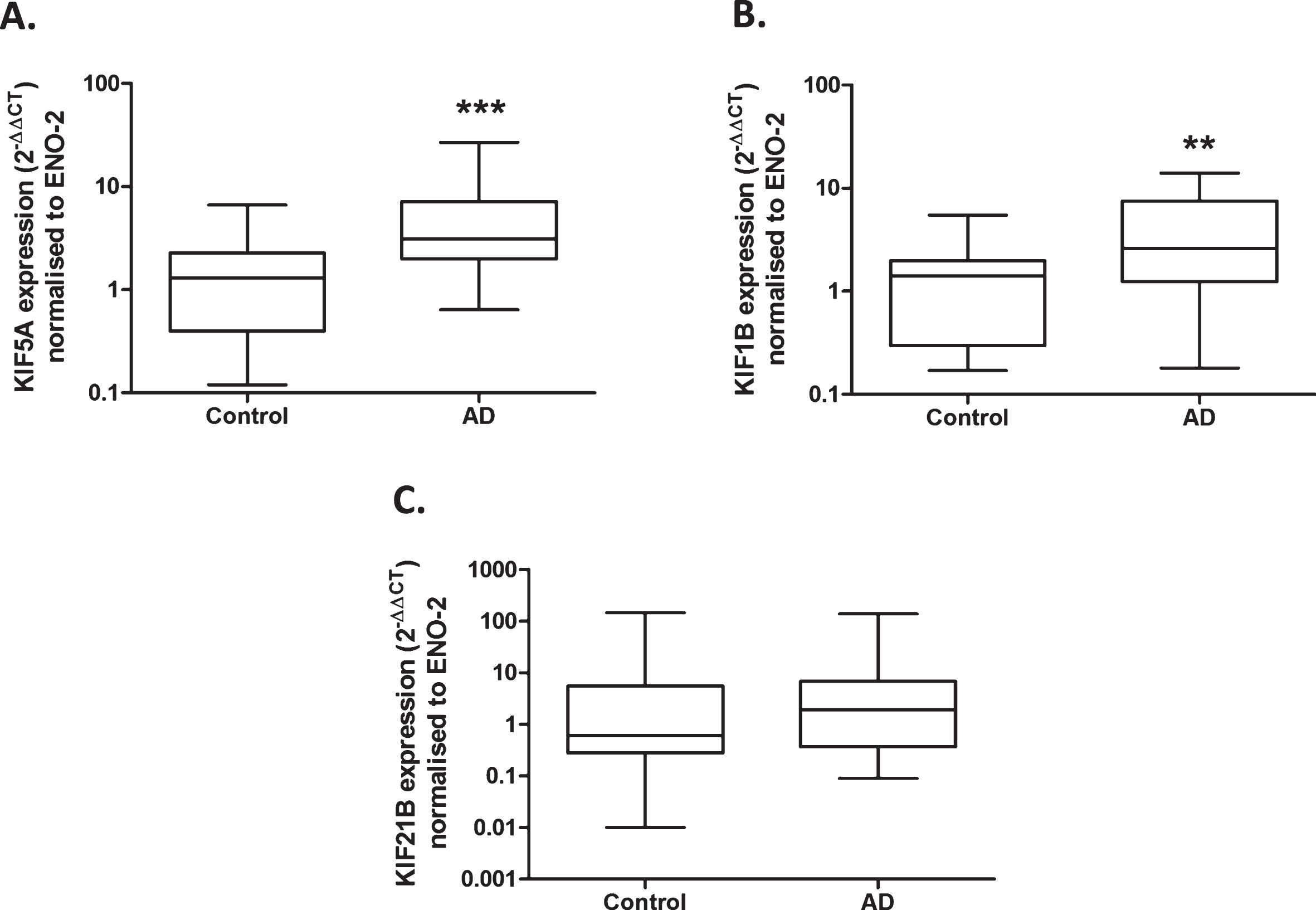
Upregulated gene expression of anterograde kinesin motors in Alzheimer’s disease. Quantitative real-time PCR performed with cDNA samples obtained from homogenized brain frontal lobe sections show a significant increase in KIF5A mRNA expression in AD cases (n = 23) compared to control (n = 22), when normalized to ENO-2 (A). KIF1B mRNA expression is significantly increased in AD cases (n = 23) compared with control (n = 22), when normalized to ENO-2 (B). KIF21B mRNA expression is not significantly different between control (n = 21) and AD cases (n = 23), when normalized to ENO-2 (C). Results expressed as median, IQR and min/max quartile. Statistical test used: two-tailed Mann-Whitney; ** p < 0.01, *** p < 0.001. AD, Alzheimer’s disease, ENO-2, enolase-2, IQR, inter-quartile range, KIF, kinesin superfamily protein.
Dot blotting
Dot blotting was performed with antibodies we had validated for specificity by western blot, as listed in Table 2. Nitrocellulose membrane was pre-soaked in 1x TBS, before placement in the 96-well Bio-Dot Microfiltration manifold (Biorad). The manifold was assembled according to the manufacturer’s protocol. Frontal lobe protein homogenates were diluted (1 : 75) with 1x TBS, and 100μL of each sample (49 AD and 46 control) was transferred by microfiltration for 90 min, including a blank TBS control well. In order to maximize the sample size available on the 96-well manifold, technical replicates were not used. The nitrocellulose membrane was subsequently removed from the manifold for blocking, antibody incubation and chemiluminescence visualization, as per the western blotting protocol. Densitometric analysis of protein dots was performed using Image Labtrademark 5.0 software (Biorad). Integrated density values were expressed relative to the neuronal control protein NeuN.
Tissue preparation for soluble and insoluble (guanidine-extractable) Aβ measurements
Approximately 200 mg of frontal tissue was homogenized in TBS extraction buffer as previously described [31]. In brief, homogenates were centrifuged at 20, 817 g for 15 min at 4°C and the supernatant (soluble fraction) stored at –80°C until use. The remaining pellet was homogenized in 6.25 M guanidine HCl (50 mM Tris/HCl, pH 8.0) and incubated for 4 h at 25°C, followed by centrifugation at 20, 817 g for 20 min, at 4°C. The supernatant (guanidine-extractable fraction) was stored at –80°C until use.
Enzyme-linked immunosorbent assay (ELISA)
Sandwich ELISA was used to measure total Aβ in the soluble and insoluble (guanidine-HCl-extractable) fractions of the homogenates, as previously described [32]. Monoclonal anti-Aβ (4G8 clone, raised against amino acids 18–22; Millipore; Watford, UK), was used for the capture step and biotinylated anti-human Aβ monoclonal antibody (10H3 clone) (Thermo Fisher Scientific; Northumberland, UK), for the detection step.
Statistical analysis
Univariate mRNA and protein analysis was carried out using GraphPad Prism5trademark (GraphPad Software Inc.; San Diego, USA). Data normality was tested using the Shapiro-Wilk test. Unpaired t-tests or non-parametric Mann-Whitney tests, as appropriate, were used to compare mRNA and protein data between the AD and control cohorts. Parametric Pearson’s or non-parametric Spearman’s correlation was used to interpret any relationship between proteins. One-way ANOVA with post-hoc Bonferroni was used to analyze KIF data categorized by Braak score. A multiple regression model (STATA v12; StataCorp LLC; Texas, USA) was used to analyze mRNA and protein expression in relation to disease, patient age of death, tissue post-mortem delay and sex. Where necessary, data was transformed to normality before performing regression analysis. For all tests, values of p < 0.05 were considered statistically significant.
RESULTS
Cohort variances
The 49 AD cases used ranged from 54–98 y (mean = 81 y, SD = 9 y) and the 46 control cases used from 43–95 y (mean = 79 y, SD = 11 y). In keeping with AD incidence, there was a higher proportion of female cases in the AD group (61.2%) compared with male (38.8%) and roughly an even gender split in control cases (female: 45.7%; male 54.3%). There was no significant difference in tissue post-mortem delay between AD (mean = 40 h, SD = 21 h) and control samples (mean = 43 h, SD = 38 h) (two-tailed Mann-Whitney; p = 0.63; Table 1). Multiple regression analysis revealed a significant effect of post-mortem delay (PMD) on mRNA expression (Table 3). Specifically, NeuN mRNA expression correlated inversely with tissue post-mortem delay (n = 47, Spearman r = –0.48, p = 0.00). Subsequent analysis excluded cases with PMD > 72 h. After exclusion, PMD still influenced NeuN mRNA expression (n = 43, Spearman r = –0.45, p = 0.00) but there was no effect of PMD on NeuN protein expression (n = 76, Spearman r = –0.11, p = 0.23). Therefore, KIF mRNA expression was normalized to an alternative neuronal house-keeping gene, neuron-specific enolase (ENO-2). There was no effect of PMD on ENO-2 mRNA expression (n = 42, Spearman r = 0.26, p = 0.09).
Multiple regression analysis of cohort variables on KIF mRNA expression
Significant correlations highlighted in bold. ENO-2, enolase-2; KIF, kinesin superfamily protein; NeuN, neuronal nuclei.
Upregulation of KIF genes in AD
There was a 4-fold increase in KIF5A mRNA relative to ENO-2 mRNA in AD compared to controls (p < 0.001; Fig. 1A). There was also a 3-fold increase in KIF1B mRNA in AD cases (p < 0.01; Fig. 1B). There was no significant difference in KIF21B mRNA expression between control and AD cases (p = 0.32; Fig. 1C). Multi-regression analysis showed no effect of patient age of death, tissue post-mortem delay or sex on KIF mRNA expression, when normalized to ENO-2 (Table 3).
Elevated KIF5A, KIF1B and KIF21B protein in AD
Immuno dot-blot revealed significantly increased KIF5A protein in AD compared to control tissue, after adjustment for NeuN content (p < 0.05; Fig. 2A). There were also significant increases in KIF1B (p < 0.001; Fig. 2B) and KIF21B protein (p < 0.001; Fig. 2C), after adjustment for NeuN. Multi-variate analysis showed no significant effect of tissue postmortem delay or sex on KIF protein expression but there was a significant effect of aging on KIF1B protein expression (Table 4). In order to establish whether increased KIF expression is an early event in AD pathogenesis, KIF expression was also sub-categorized according to Braak stage, ranging from 0–6. One-way ANOVA with post-hoc Bonferroni did not reveal significant differences between the groups (Supplementary Table 1).
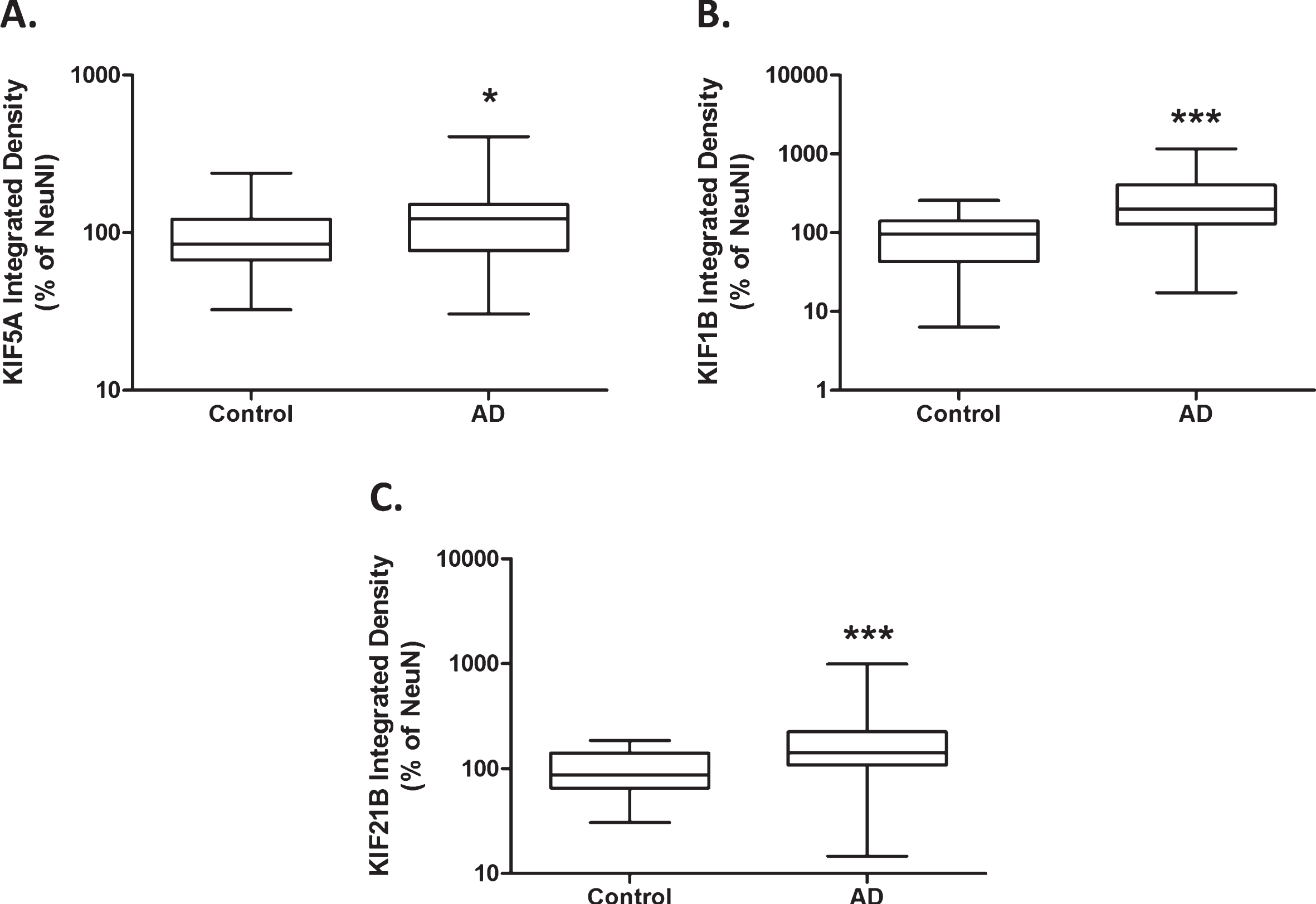
Increased anterograde kinesin motor protein expression in Alzheimer’s disease. Protein levels derived from immuno dot-blot show a significant increase in anterograde kinesin motor protein KIF5A expression in AD cases (n = 43), compared with control (n = 35), when normalized to NeuN (A). KIF1B protein expression is significantly increased in AD cases (n = 42), compared with control (n = 33), when normalized to NeuN (B). KIF21B protein expression is significantly increased in AD cases (n = 44), compared with control (n = 36), when normalized to NeuN (C). Results expressed as median, IQR and min/max quartile. Statistical test used: two-tailed Mann-Whitney; * p < 0.05, *** p < 0.001. AD, Alzheimer’s disease; IQR, inter-quartile range; KIF, kinesin superfamily protein; NeuN, neuronal nuclei.
Multiple regression analysis of cohort variables on protein expression
Significant correlations highlighted in bold. Aβ, amyloid-β, AD, Alzheimer’s disease, AβPP, amyloid-β protein precursor, KIF, kinesin superfamily protein, PHF, paired helical filament.
KIF5A protein correlates inversely with hyperphosphorylated tau in AD brains
As expected, we found a significant increase in hyperphosphorylated tau in AD compared to controls, adjusted for NeuN (p < 0.001; Fig. 3). This was replicated in multi-variate analysis, however, there was a significant influence of patient age on tau expression (p < 0.05, Table 4). Univariate analysis showed no correlation between KIF5A expression (adjusted for GAPDH [33]) and hyperphosphorylated tau (adjusted for NeuN) in control (Pearson r = –0.30, p = 0.09) or AD cases (Spearman r –0.27, p = 0.10; Table 5). Due to the influence of age, additional multi-variate analysis was performed which revealed a significant effect of KIF5A protein on hyperphosphorylated tau expression in AD cases (p = 0.04, Table 6). Both univariate and multi-variate analysis showed no significant correlations between tau and KIF1B or KIF21B (Tables 5 and 6).
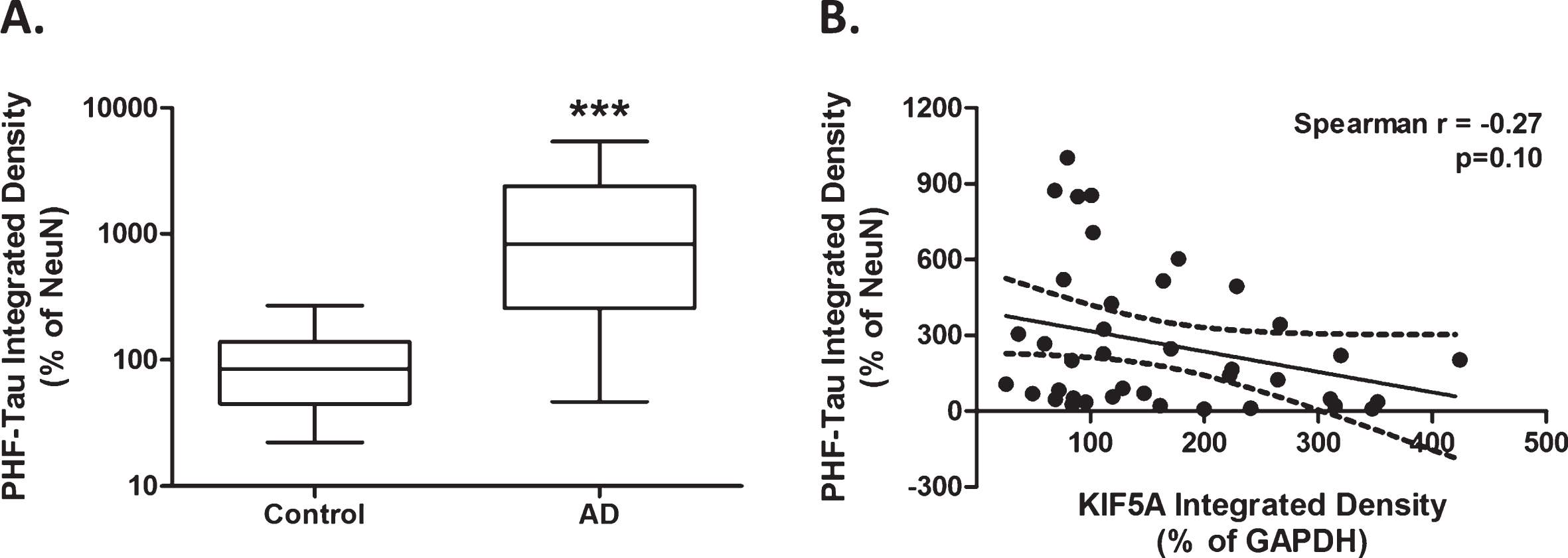
Elevated hyperphosphorylated tau protein expression in AD cases. Protein levels derived from immuno dot-blot show a significant increase in PHF-tau expression in AD cases (n = 42) compared with control (n = 35), when normalized to NeuN (A). Univariate analysis shows no significant correlation between KIF5A and PHF-Tau levels in AD cases (n = 42; B). Results expressed as median, IQR and min/max quartile. Statistical test used: two-tailed Mann-Whitney (A). Correlation represented as line of best fit±95% CI (B). *** p < 0.001. AD, Alzheimer’s disease; CI, confidence interval; IQR, inter-quartile range; KIF, kinesin superfamily motor protein; NeuN, neuronal nuclei; PHF, paired helical filament.
Correlation analysis of kinesin superfamily protein with proteins linked to Alzheimer’s disease
Significant correlations highlighted in bold. Aβ, amyloid-β; AD, Alzheimer’s disease, AβPP, amyloid-β protein precursor; KIF, kinesin superfamily motor protein; PHF, paired helical filament.
Multiple regression analysis of KIF, age, and tau protein expression
Significant correlations highlighted in bold. AD, Alzheimer’s disease; KIF, kinesin superfamily motor protein; PHF, paired helical filament.
KIF5A protein correlates inversely with AβPP and soluble Aβ in AD
AβPP protein level (adjusted for NeuN) was significantly elevated in AD tissue compared with control in univariate analysis (p < 0.05; Fig. 4A). However, this increase was not significant in subsequent multi-variate analysis, considering patient age of death, tissue PMD, and sex (p = 0.15, Table 4). There was a significant inverse correlation between KIF5A (adjusted for GAPDH) and AβPP protein level (adjusted for NeuN) in AD cases (Spearman r = –0.53, p < 0.001; Fig. 4B) and control cases (Spearman r = –0.42, p < 0.05; Table 5). As expected, there was a significant increase in insoluble Aβ levels in AD cases compared with control (two-tailed Mann-Whitney, p < 0.001), which was consolidated in multi-variate analysis (Table 4). There was no correlation between KIF5A expression and insoluble Aβ in AD cases (Pearson r = 0.05, p = –0.08; Table 5). Soluble Aβ was elevated in AD cases but not to significance (p = 0.06; Fig. 4C), as verified in multi-variate analysis (p = 0.05; Table 4). Like AβPP, soluble Aβ levels correlated inversely with KIF5A in AD cases (Pearson r = –0.49, p < 0.05; Fig. 4D). Previous studies have suggested that the level of soluble Aβ tends to fall with increased deposition of insoluble Aβ [34, 35]. However, KIF5A level did not correlate with the ratio of soluble: insoluble Aβ in AD cases (Spearman r = –0.35, p = 0.09; Table 5). Univariate analysis showed no association between KIF21B or KIF1B with AβPP, soluble Aβ, insoluble Aβ and soluble:insoluble Aβ in control or AD cases (Table 5). Considering the effect of age on KIF1B expression, multi-variate analysis was also performed which showed no significant correlation between KIF1B and AβPP, soluble Aβ and insoluble Aβ, which was not affected by age (Supplementary Table 2).
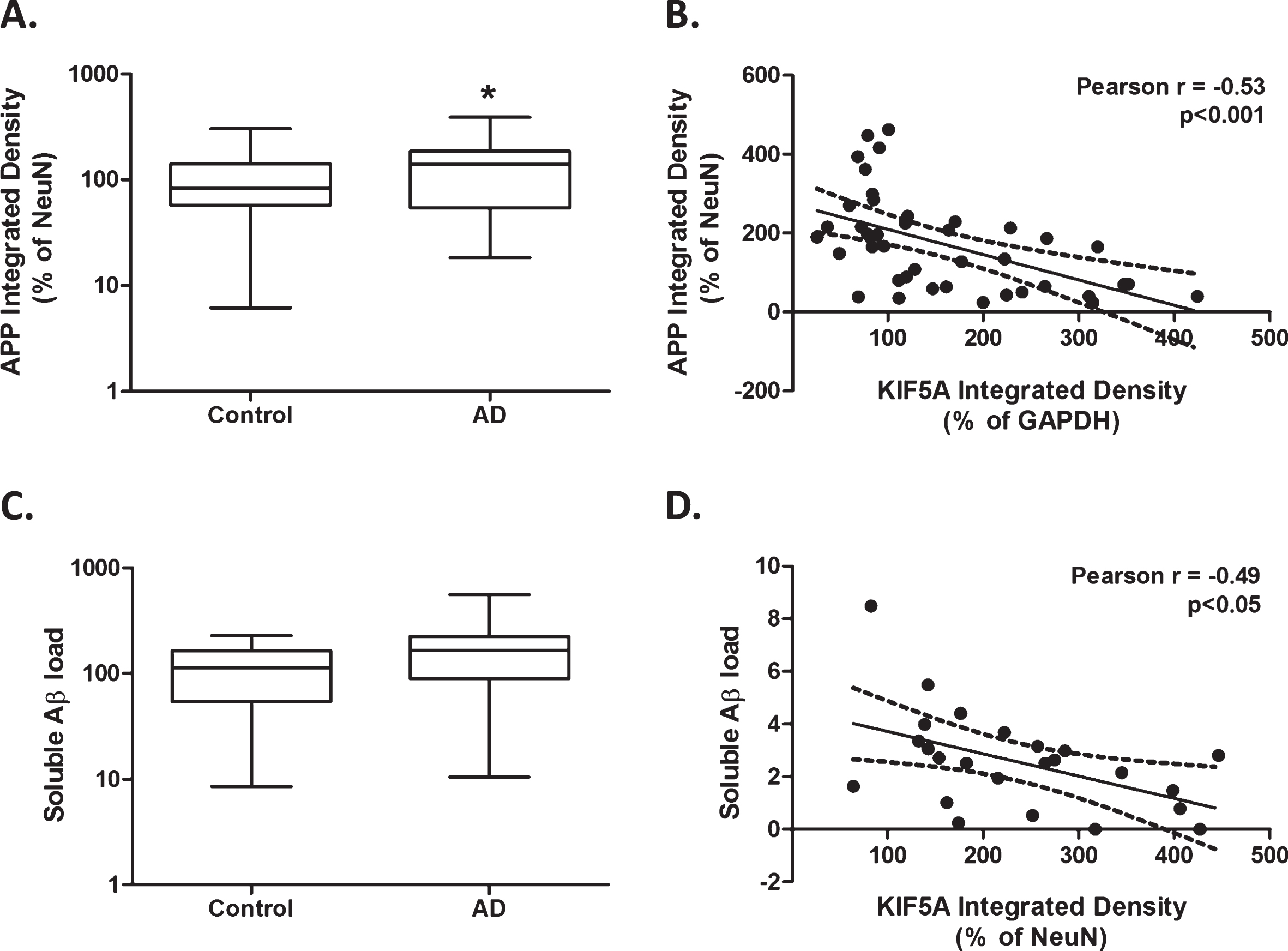
Higher kinesin motor KIF5A protein levels correlate with lower levels of amyloid precursor protein and soluble Aβ in AD. Protein levels derived from immuno dot-blot show a significant increase in AβPP expression in AD cases (n = 45) compared with control (n = 36), when normalized to NeuN (A). Significant inverse correlation between KIF5A protein levels normalized to GAPDH and AβPP protein levels, normalized to NeuN in AD (n = 41; B). Protein levels obtained via ELISA show no significant difference in soluble Aβ protein expression in AD cases (n = 42) compared with control (n = 22; C). Significant inverse correlation between KIF5A protein levels normalized to NeuN and soluble Aβ protein levels in AD (n = 24; D). Results expressed as median, IQR and min/max quartile. Statistical test used: two-tailed Mann-Whitney (A and C). Correlations represented as line of best fit±95% CI (B and D). * p < 0.05, *** p < 0.001. Aβ, amyloid-β; AD, Alzheimer’s disease; AβPP, amyloid-β protein precursor; CI, confidence intervals; IQR, inter-quartile range; KIF, kinesin superfamily protein; NeuN, neuronal nuclei.
DISCUSSION
Dysregulated KIF expression has been investigated in relation to several neurodegenerative diseases but remains relatively little studied in AD [29, 36]. We have found increased KIF gene and protein expression in AD, and significant inverse correlations between KIF5A expression and AβPP and soluble Aβ in AD.
KIF5A exists predominantly as a tetramer comprising two dimerized kinesin heavy chains, and two kinesin light chains (KLC1 and KLC2) attached at the tail domain of the KHCs. Studies in mice have shown KIF5A knock-out is neonatal lethal and post-natal targeting of the gene results in reduced axon caliber, eventual axon loss, and hind-limb paralysis [9]. KIF5A is believed to transport several cargoes through association with KLCs, including AβPP, phosphorylated NFs, and SNARE complex components; SNAP-25 and syntaxin-1b [17]. It has been suggested that genetic variability in the KLC1 gene may influence the development of AD [20]. In mice, deletion of the KLC1 subunit leads to early selective defects of axonal transport of several cargoes including AβPP, NF, and tau aggregates, causing cytoskeletal disorganization [37].
We found increased KIF5A protein in AD to be associated with elevated levels of the corresponding transcript. It is possible that KIF5A is upregulated as an adaptive response in an attempt to clear or circumvent large protein aggregates within the neuron or to compensate for potential reduced activity of other motors. Elevated KIF5A mRNA has also been found in brain tissue from demented compared to non-demented patients with Parkinson’s disease [38]. Univariate analysis showed a significant increase in AβPP levels in AD compared with control. However, this effect was lost in multi-variate analysis of the cohort, in keeping with previous studies [39, 40]. KIF5A protein levels correlated inversely with that of AβPP, as previously reported in MS white matter [13]. AβPP is a type I transmembrane protein with suspected roles in cell adhesion, regulation of gene expression, and iron export [41]. AβPP transport by KIF5A is thought to be mediated by KLC association with AβPP-containing vesicles [18]. The subcellular distribution of AβPP plays a critical role in its metabolism, including the cleavage by β- and γ-secretases that generates pathogenic Aβ peptides [35, 42–52 , 35, 42–52]. Maintenance of neuronal transport of AβPP by upregulation of KIF5A may help to maintain normal metabolism of AβPP (and thereby neuronal viability) for longer than would otherwise be the case. In support of this, although soluble Aβ levels were higher in AD than control tissue, the levels correlated inversely with that of KIF5A. KIF5A level was not related to the ratio of soluble: insoluble Aβ, suggesting that KIF5A does not significantly influence the deposition of Aβ. However, we hypothesize that upregulation of KIF5A may temporarily ameliorate the damaging effects of abnormal intracellular protein aggregates through maintained intraneuronal transport.
NFTs form through aggregation of hyperphosphorylated tau [53]. Multi-variate analysis of our cohort revealed levels of hyperphosphorylated tau declined with age. This is likely to be influenced by many confounding variables such as diet, genetics, and lifestyle [54]. However, taking aging into account, we found KIF5A levels correlated inversely with hyperphosphorylated tau levels in AD cases. This again supports the hypothesis that KIF5A upregulation is crucial in maintaining intraneuronal transport. However, the effectiveness of KIF5A-mediated transport in AD still needs to be determined. Studies have reported that pathogenic forms of tau inhibit axonal transport through activation of the protein phosphatase 1/glycogen synthase kinase 3 (PP1/GSK3) pathway [55], which is believed to cause dissociation of KIF5A from its cargo through phosphorylation of KLC [29].
KIF1B mRNA and protein levels were higher in AD than control tissue. KIF1B has two splice variants, KIF1Bα and KIF1Bβ, which exist as KHC monomers. KIF1Bα transports mitochondria, synaptic scaffolding molecules (SCAM) and post-synaptic density (PSD) proteins PSD-95 and PSD-97. KIF1Bβ transports synaptic vesicle precursors including synaptotagmin, synaptophysin and SV2. In humans, a missense mutation in the KIF1Bβ gene is linked to CMT-2A [27], characterized clinically by weakness and atrophy of distal muscles, depressed or absent tendon reflexes and mild sensory loss. The kif1bβ mutation results in a glutamine to leucine substitution in the ATP-binding site of the kif1bβ motor domain, which causes a reduction in microtubule-ATPase activity and consequent reduction in the transport of cargoes. Impaired delivery of synaptic vesicle precursors to axons and nerve terminals contributes to progressive dysfunction of peripheral neurons [56]. Because of their overlapping cargoes, a level of functional redundancy is thought to exist between KIF1B and KIF5A. Campbell et al. [15] studied functional redundancy of KIFs in zebrafish and found that kif1b overexpression cannot compensate for loss of kif5a-mediated mitochondrial transport but suggested a dual function in maintaining peripheral sensory neuronal function through the transport of non-mitochondrial cargoes. These non-mitochondrial cargoes are most likely to be synaptic proteins. Indeed, rodent studies have suggested alternative roles for KIF5A and KIF1B in modulating mitochondrial motility, suggesting KIF1B overexpression reduces mitochondrial transport, whereas KIF5A overexpression increases motility [57]. We had considered the possibility that elevated KIF5A might partly be a physiological compensatory response to reduced KIF1B. We found both KIF5A and KIF1B to be elevated in AD, however, further investigation of family isoforms, in particular KIF1Bα and KIF1Bβ, could help elucidate potential subunit redundancy. The precise affinity of KIF5A for cargoes such as AβPP and the degree of functional redundancy between axonal motors from different sub-groups remains an open question [58].
The anterograde motor protein KIF21B was increased in AD compared with control tissue. KIF21B is enriched in the dendrites and transports γ2-subunit-containing GABAA receptor vesicles. Altered expression of KIF21B may be involved in the regulation of receptor density that mediates GABAergic synaptic plasticity [28]. Kreft et al. [59] found that KIF21B mRNA was increased in early-onset AD (≤62 y at the time of death) and that the level correlated with shorter disease duration, and increased disease severity as assessed by neuropathology. In our study, the elevation in KIF21B mRNA in AD was not statistically significant. However, the mean age at death in our AD cohort was 81 y, and Kreft et al. did not find any increase in KIF21B mRNA in an older AD cohort (≥72 y atdeath).
In summary, we have found upregulation of three KIFs in AD. It remains to be determined whether upregulation is an adaptive response that helps to maintain intraneuronal transport and stabilize axonal structure in AD or whether it contributes to neurodegeneration.
Footnotes
ACKNOWLEDGMENTS
We thank the South West Dementia Brain Bank (SWDBB) for providing brain tissue for this study. The SWDBB is part of the Brains for Dementia Research program, jointly funded by Alzheimer’s Research UK and the Alzheimer’s Society, and is supported by BRACE (Bristol Research into Alzheimer’s and Care of the Elderly) and the Medical Research Council.