
Abstract
Background:
During the twentieth century, frontotemporal dementia (FTD) was often misdiagnosed, confused with Alzheimer’s disease or psychiatric disorders, jeopardizing care and research.
Objective:
To analyze the FTD genes in the DNA samples of patients belonging to families clinically classified as probable Alzheimer’s disease (FAD) in the early 1990s and not carrying mutation in the three main genes linked to FAD (Presenilin 1, Presenilin 2, and Amyloid precursor protein).
Methods:
The genetic screening was performed on 63 probands diagnosed as FAD before the early 2000s.
Results:
Four patients out of the 63 studied (4/63, 6.3%) resulted as carrying four different GRN genetic variations: p.T272SfsX10, p.R110X, p.C149LfsX10, and p.W304C. The first two mutations (p.T272SfsX10, p.R110X) are the most frequent ones in Italy in FTD patients; the latter two (p.C149LfsX10 and p.W304C) are not described in the scientific literature.
Conclusion:
Our data suggest that it can be important to re-examine FAD patients diagnosed when the FTD spectrum was not well recognized and the causative FTD genes had not yet been identified. Moreover, we propose initially analyzing genes associated with the first form of suspected dementia and, if the results are negative, studying genes implicated in the other form of dementia.
INTRODUCTION
Frontotemporal dementia (FTD) is considered the second most frequent cause of dementia after Alzheimer’s disease (AD). The disease was described for the first time at the end of the 1800s, as Pick disease; however during the twentieth century, it was often misdiagnosed, confused with AD or psychiatric disorders, jeopardizing care and research [1]. Presently, it is known that FTD is a genetically complex disorder with multiple genes contributing to the disease [2]. The first gene correlated with the disease was discovered in 1998 when mutations in the microtubule-associated protein tau (MAPT) gene were identified in a portion of FTD and parkinsonism families [3, 4] previously linked to chromosome 17q (FTDP-17) [5–7]. The described FTDP-17 families with linkage to chromosome 17q21, but negative for MAPT mutations, made it possible in 2006 to identify the Granulin gene (GRN) located 2 centimorgans from the MAPT [8, 9]. GRN mutations are responsible for 5–20% of familial and 1–5% of sporadic FTD cases [10]. To date, more than ten different genes have been related to the disease: mutations in MAPT and GRN genes and the presence of the hexanucleotide expansion repeats in the open reading frame of chromosome 9 (C9orf72) cover 60% of familial FTD cases, while <5% of the familial cases are caused by mutations in valosin-containing protein (VPC), chromatin-modifying 2B (CHMP2B), TARDNA binding protein 43 encoding gene (TARDBP), fused in sarcoma gene (FUS), sequestosome 1 (SQSTM1), ubiquilin (UBQLN), optineurin (OPTN), triggering receptor expressed on myeloid cells 2 (TREM2), coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10), and tank-binding kinase 1 (TBK1) [2].
The aim of the present study was to analyze the MAPT, C9orf72, and GRN genes in the DNA samples of patients belonging to families clinically classified as probable AD (FAD) in the early 1990s, not carrying mutation in the three main genes linked to FAD: Presenilin 1 (PSEN1), Presenilin 2 (PSEN2), and Amyloid precursor protein (APP).
METHODS
The genetic screening was performed on 63 probands diagnosed as FAD before the early 2000s (44 female 69.8%, age at onset 64.2±10.2 years).
The diagnosis of probable AD was performed following the clinical criteria of the National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer’s Disease and Related Disorders Association (NINCDS/ADRDA) criteria [11].
The study protocol was approved by the local ethics committee and conducted in accordance with the provisions of the Helsinki Declaration.
Total DNA was isolated from peripheral blood using standard methods. All the coding exons and the intron/exon boundaries of MAPT, GRN were PCR amplified with primers designed using Primer3 software, and the analysis was performed using High Resolution Melting Analysis (HRMA). Samples having an unusual melting profile were sequenced using BigDye TerminatorTM protocol on an automated 310 ABI PRISM Genetic Analyzer (Applied Biosystem).
The C9orf72 repeat expansion was searched using the repeat-primed PCR and sequencing [12]; the characteristic stutter amplification pattern was considered as indication of pathogenic repeat expansion (>30 repeats).
RESULTS
In our study, 63 probands diagnosed as FAD before the early 2000s were screened, and they were not carrying either mutations in MAPT gene or the C9orf72 repeat expansion.
Four patients out of the 63 studied (4/63, 6.3%) were found to be carrying four different GRN mutations: p.T272SfsX10, p.R110X, p.C149LfsX10, and p.W304C (Fig. 1). The first two mutations, p.T272SfsX10 (Fig. 1A) and p.R110X (Fig. 1B), are the most frequent ones in Italy in FTD patients. The latter two, p.C149LfsX10 (Fig. 1C) and p.W304C (Fig. 1D), are not described in scientific literature.
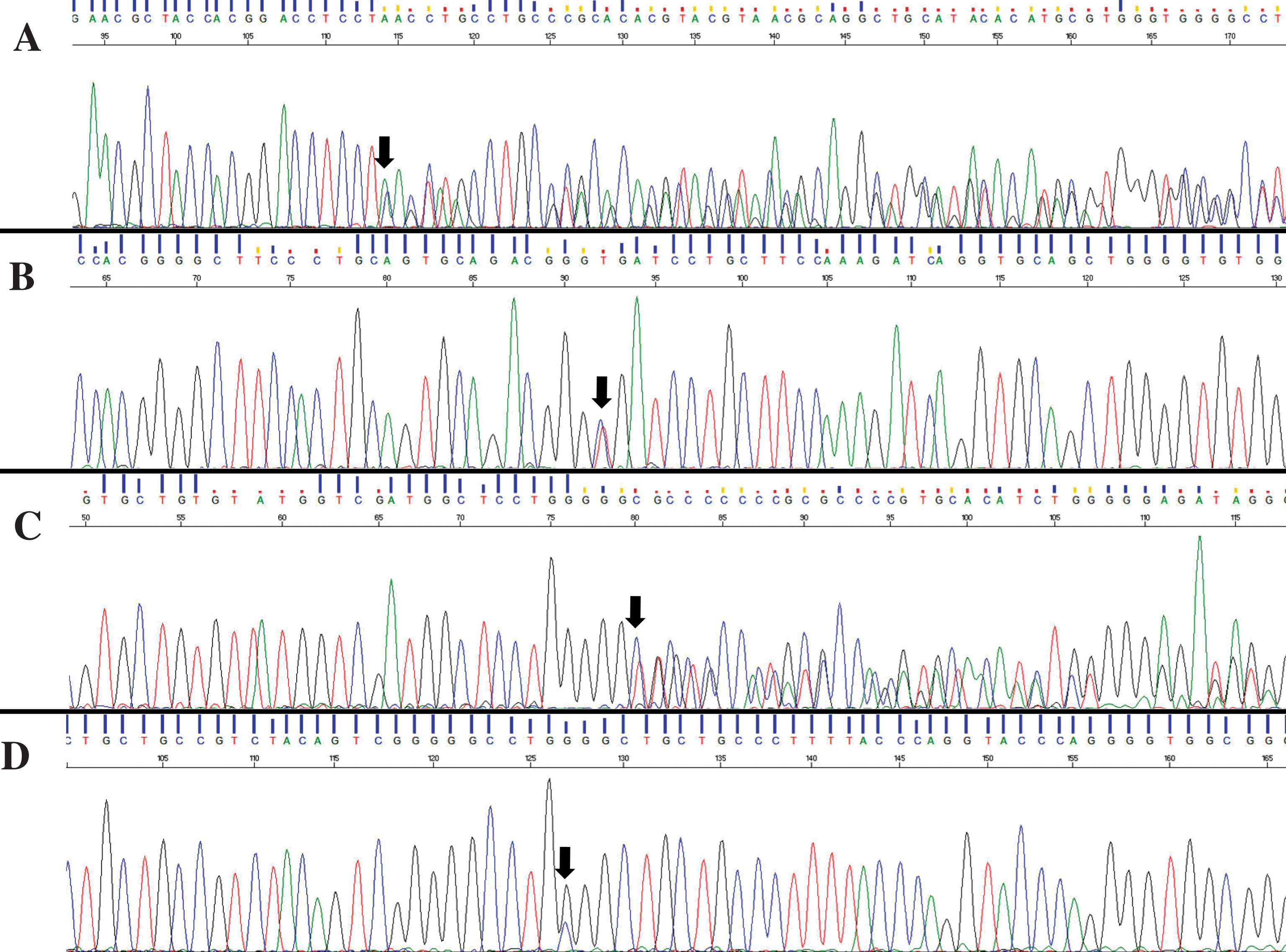
Sequencing chromatograms of the described GRN variations: p.T272SfsX10 (A), p.R110X (B), p.C149LfsX10 (C), and p.W304C (D).
The p.T272SfsX10 mutation (Fig. 1A) was identified in a female patient with an age at onset of 64 years in 1991. The neuropsychological tests conducted revealed a prominent initial episodic memory impairment, depression and apathies that were the principal cause of compromised daily living activities; in 1992 the final AD diagnosis was made. Her familial history was strongly suggestive for cognitive impairment, although reliable medical records were available only for a few relatives. Her father died at the age of 62 years, due to complications in cerebral arteriosclerosis and cognitive impairment, and her brother and sister died at 46 and 60 years of age, respectively, with a diagnosis of cognitive impairment. Moreover, her paternal aunt received a diagnosis of cognitive impairment at the end of the 1980s.
The patient carrying the p.R110X (Fig. 1B) was a 63-year-old woman who underwent neurological examination in an already advanced stage of the disease and was not able to perform neuropsychological assessments or other tests in 1996. Family history for cognitive impairment was signaled. Her father was referred to as having been affected by dementia. Two already deceased paternal aunts were affected by cognitive impairment. Unfortunately, it was not possible to analyze any family member.
Here we present in detail the case reports for the two unknown genetic variants: p.C149LfsX10 (Fig. 1C) and p.W304C (Fig. 1D).
Case report carrying the p.C149LfsX10
The p.C149LfsX10 is a novel loss-of-function mutation (according to the validated haploinsufficiency mechanism of the known GRN mutations, thus considered pathogenetic) identified in a female patient with an age at onset of 62 years. The first neuropsychological evaluation was carried out in 1997 at the age of 64 years, two years after onset of symptoms. Spontaneous speech was characterized by slow rate, with pauses due to anomic problems, repetitive prosody and grammatical errors such as deletions or substitution of morphemes. Deficit of verbal fluency was detected both for phonemic and semantic stimuli, and sentence repetition resulted impaired. Other cognitive domains were normal. Computed axial tomography (CT) resulted negative for vascular lesions. In 1997, a clinical diagnosis of a focal variant of AD was made and the genetic analysis on the three candidate AD genes was negative.
After two years, speech difficulties had progressed and were associated with buccofacial apraxia and ideomotor apraxia; however, functional autonomy in everyday life was still preserved. At 68 years of age, a formal evaluation was not applicable due to severe language deficits, characterized by absence of spontaneous speech and severe comprehension impairment; the patient was dependent for everyday life activities.
A family history of dementia was positive (FLO 46, Fig. 2). In fact, a generic diagnosis of “dementia” had been made in her mother (I-2) and brother (II-2). Moreover, up to now, her son (III-1) at the age of 63 years shows the first symptoms of apathy, cognitive impairment, and aphasia. It was not possible to verify the presence of the variant in the affected family members as I-2 and II-2 are deceased and III-1 did not give his consent. The variant was found in neither ExAC [13] nor 1000G [14] data sets (which contain exomes from 60,706 unrelated individuals); moreover, a free online bioinformatics prediction software (MutationTaster [15]) was used in order to evaluate the GRN mutation pathogenicity. In silico analysis revealed that the p.C149LfsX10 could be deleterious with a high probability (MutationTaster-Disease causing, score probability 1).
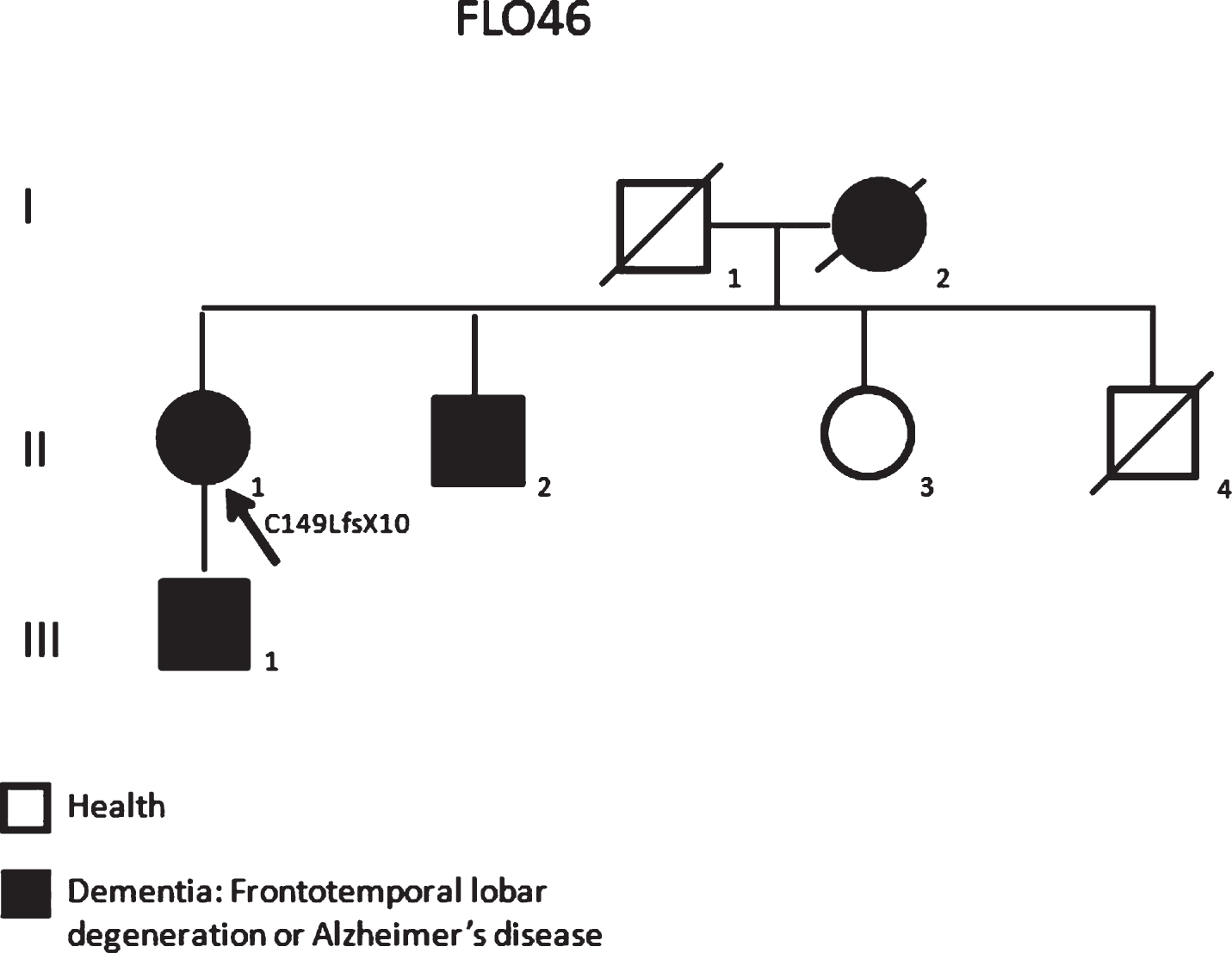
The pedigree for the studied family FLO46 carrying the p.C149LfsX10 mutation showing a family history of dementia. (Arrows in the pedigrees showed probands).
Case report carrying the p.W304C
The p.W304C is a novel missense variation identified in a female patient with an age at onset of 65 years diagnosed as probable FTD in 2012. The patient complained of mild behavioral symptoms (social retirement) and memory problems starting in 2011. A brain magnetic resonance imaging (MRI) study highlighted diffuse cortical atrophy, more evident in anterior lobes. The neuropsychological evaluation disclosed a dysexecutive syndrome with impaired working memory and verbal fluency. Left hand apraxia and dysdiadochokinesia were also evident. In the following years, the patient’s clinical picture progressively deteriorated. At the last follow-up visit in September 2014, she was able to answer only very simple questions, was severely dysarthric and dysphagic to liquids. Standing up and walking were impossible and she was completely dependent on her relatives for all daily activities.
Collecting information about the family, we found members already described and diagnosed as FAD (FLO16) in the early 1990s (Fig. 3). Her mother (II-2) as well as maternal grandmother (I-2) and one aunt (II-6) had been diagnosed with “dementia”. Moreover, two of the patient’s brothers (III-7 and III-8) received a diagnosis of AD a few years later and their blood samples, analyzed for the three known FAD genes, were negative. Thus, we verified the presence of the novel missense variation p. W304C in GRN in all members of the family. The same variant is present in the two affected brothers (III-7, age at onset 50 years; III-8, age at onset 55 years) but also in one healthy sister (III-4, 79 years old), whereas the two healthy sisters (III-5 and III-6, 69 and 62 years old, respectively) and one healthy brother (III-9, 77 years old) resulted negative.
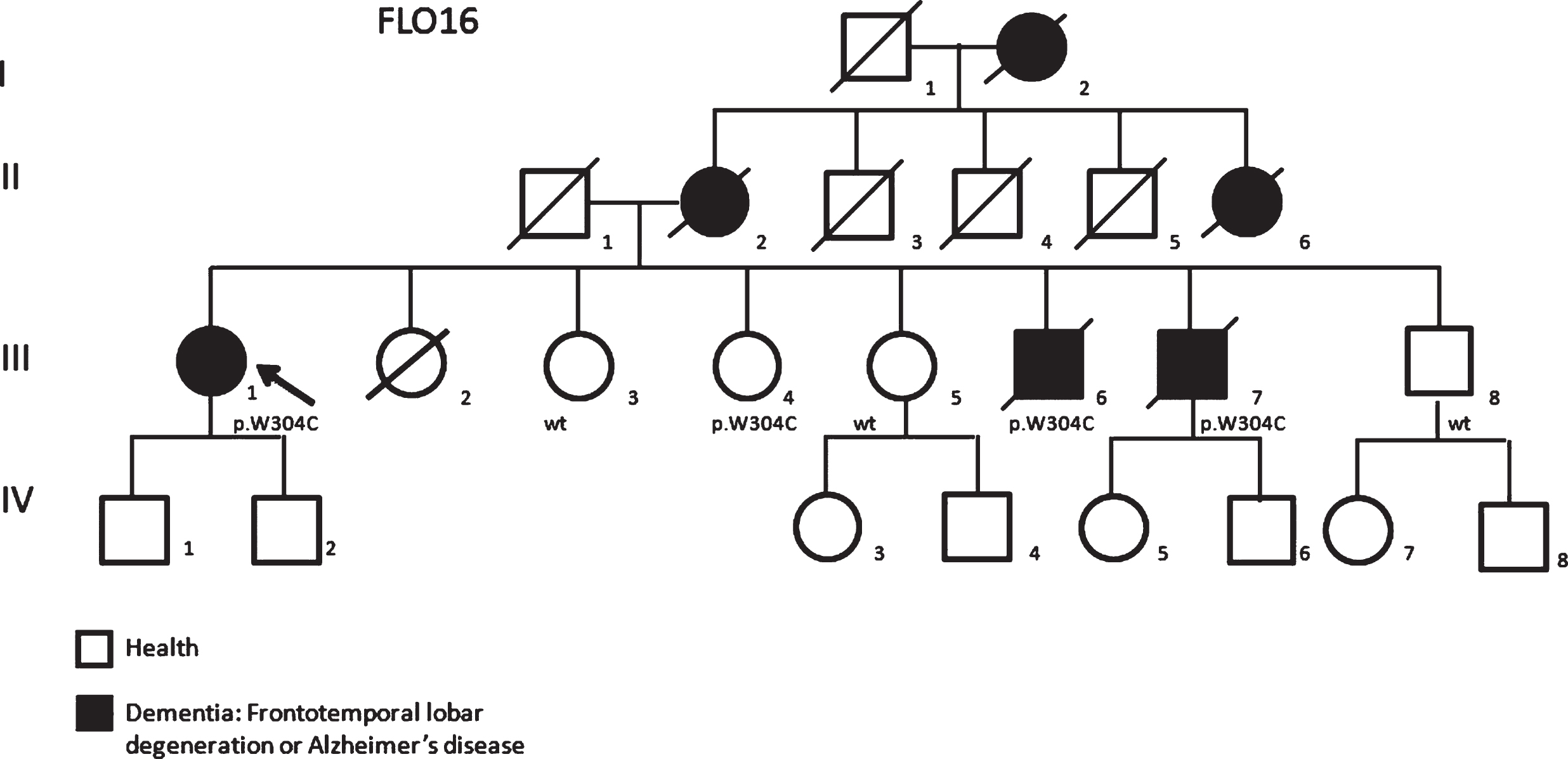
The pedigree of the family FLO16 carrying the p.W304C variant showing a clear family history of dementia (Arrows in the pedigrees showed probands).
The variant was found in neither ExAC [13] nor 1000G [14] data sets and its effect on the function of the protein has been estimated using free online bioinformatics prediction tools: MutationTaster [15], MUpro [16] and PolyPhen2 [17]. In silico analysis revealed that the modified amino acid was conserved in all the analyzed species, thus the p.W304C variant could be deleterious (MutationTaster-Disease causing, score probability-0.99; MUpro-Decrease stability, score –0.59; PolyPhen2-Possibly damaging, score 1).
DISCUSSION
In the early 1990s, FTD was a neurodegenerative dementia that was difficult to clinically distinguish from AD [18], which paved the way for intense debate about testing the capacity of the current NINCDS-ADRDA criteria [11] in clinical differential diagnosis. Varma and colleagues, in 1999, assessed the capability of the criteria to accurately distinguish AD from FTD in a series of pathologically proven cases and thus suggested revising them in order to improve their specificity as they were not able to provide a differential clinical diagnosis between the two diseases [19]. Today, the clinician finds support from integration of clinical information, validated biomarkers, genetic analysis, and brain imaging techniques such as Amyloid PET and the FDG PET, key aspects which allow discrimination between different disorders. However, the differential diagnosis of dementia forms is still difficult today due to the clinical symptomatology overlapping and for this reason an AD-FTD spectrum has been defined [20].
Moreover, in the early 1990s, the first approach to study patients with familial dementia was the genetic analysis for FAD, as FTD genes had not yet been discovered (MAPT and GRN were described in 1998 and 2006, respectively) [3, 9]. Thus, we decided to revise all cases studied in the 1990s with family history of dementia not carrying pathogenetic mutations in FAD genes.
With this approach, we identified three different GRN mutations: p.T272SfsX10, p.R110X, and p.C149LfsX10 in three patients and an unknown variant (p.W304C). The first two mutations (p.T272SfsX10, p.R110X) are the most frequently described in FTD patients in Italy, while the latter (p.C149LfsX10) is a new pathogenic mutation. In fact, the creation of a premature stop codon leads to the reduction of the GRN production which is the most common pathological mechanism.
At the end of the 1990s, cerebrospinal fluid biomarkers (associated with AD in 2003) [21] and plasma Progranulin levels (discovered in 2008) [22] were unknown and therefore in 1997, lumbar puncture and plasma analysis were not scheduled for our case (the C149LfsX10 mutation carrier).
With regards to p.W304C, the variant does not cause haploinsufficiency of the protein, but the addition of a cysteine residue inside the protein may affect the unique three-dimensional structure of the protein. The GRN protein structure requires six disulfide bridges and, among the known missense pathogenetic mutations of GRN, the mutations p.Cys521Tyr and p.Cys139Arg (affecting a cysteine residue) were demonstrated to be pathogenic, providing evidence of a consistent role of these residues in the protein structure [23, 24]. The p.W304C, like the p.Cys521Tyr and p.Cys139Arg, affects the protein conformation and could cause wrong successive proteolytic cleavages by elastase, but not a reduction of protein secretion. The presence of the missense variant in all the affected members of the family could support the hypothesis of its pathogenic role. Moreover, the GRN classic loss of function mutations penetrance is highly variable, multiple non-penetrant carriers being reported with ages older than the age at onset of the family [25], like the healthy subject carrying the variant (III-4).
Nowadays, despite the integrated use of advanced techniques (neuroimaging, biomarkers, and genetics) [26], the guidelines do not yet allow clinicians to definitely diagnose and distinguish the major forms of dementia [27]. In particular, patients with familial form of dementia with early age at onset and typical FTD clinical presentations have recently been described as carrying mutations in FAD genes [28, 29]. On the other hand, patients clinically diagnosed initially as AD resulted as carriers of pathogenic mutations in FTD genes [30, 31].
In conclusion, our data suggest that it can be important to re-examine FAD patients diagnosed when the FTD spectrum was not well recognized and the causative FTD genes had not yet been identified. The genetic approach involving the FTD genes did not directly help the selected patients get a refined diagnosis, as most of them had already died, but could have important implications for their families and mostly provides evidence that in cases of a familial form of dementia it may be important to initially analyze genes associated with the first form of suspected dementia and, if the results are negative, to study genes implicated in the other form of dementia.