
Abstract
Alzheimer’s disease and other dementias present with tau pathology. Several mouse lines with knockout of the tau-encoding Mapt gene have been reported, yet findings often differed between lines and sites. Here, we report a new tau knockout strain (tauΔex1), generated by CRISPR/Cas9-mediated genome editing of intron -1/exon 1 of Mapt in C57Bl/6J mice. TauΔex1 mice had no overt phenotype, but, in line with previous models, they showed a significantly reduced susceptibility to excitotoxic seizures, with normal memory formation in young mice. This new in vivo resource will be made freely available to the research community.
INTRODUCTION
The tau protein belongs to a family of microtubule-associated proteins (MAPs) and is found at high levels in neurons. In humans, tau is encoded by the microtubule-associate protein tau (MAPT) gene on chromosome 17, while in mice, the corresponding Mapt gene is located on chromosome 11. The MAPT mRNA is subject to alternative splicing of exons 2, 3, and 10, giving rise to 6 different isoforms found at equal levels in the adult human brain, and containing either 0, 1, or 2 amino-terminal inserts (encoded by exons 2 and 3) and 3 or 4 microtubule-binding repeats (3/4R) in its carboxy-terminal half [1]. During embryonic development in both humans and mice, 3R tau isoforms are predominant. The adult mouse brain differs from the human brain, in that only 4R tau isoforms are present shortly after birth [2, 3]. Functionally, tau contributes to dynamics of the microtubule cytoskeleton and regulates cargo transport along neuronal processes [4]. We have shown in addition that tau has important functions in regulating post-synaptic signaling processes [5–7].
Tau rose to fame when it was discovered as the major constituent of neurofibrillary tangles in Alzheimer’s disease (AD), where it undergoes aberrant hyperphosphorylation [8]. Furthermore, tau becomes hyperphosphorylated and forms intracellular deposits in a range of frontotemporal dementia (FTD) subtypes [9]. Familial forms of FTD have been linked to pathogenic mutations in MAPT [10]. Accordingly, transgenic mice expressing mutant human tau have contributed to the understanding of disease mechanism in AD and FTD [11, 12]. Conversely, tau-deficient mice (tau–/–) have been generated to study its role in disease and physiology [13]. Interestingly, tau–/– mice initially did not present with overt deficits [14, 15], possibly due to compensation by other MAPs [16]. More detailed testing revealed specific deficits in some tau–/– lines as they age [17–20], while others did not report functional impairments even at advanced ages [6, 21–23]. A critical role in the early pathogenesis of AD was established by showing that amyloid-β-induced deficits were prevented on a tau–/– background [7, 24], although one report showed enhanced pathology under similar experimental conditions [25]. It has been suggested that the conflicting findings from tau–/– mice may be due to variable genetic background and/or differences in holding and nutrition of the mice [23]. Furthermore, all tau–/– lines characterized so far have been generated by ES-cell gene targeting, which relies on the insertion of large transgenes in the coding part of the Mapt gene to disrupt its expression [13]. The insertion of large selectable marker cassettes (typically after gene trap experiments) can create phenocopy and/or unwanted confounding effects [26–28] and create inconsistent results.
Here, we report the generation of a new tau–/– resource by targeting the transcriptional start codon in exon 1 of Mapt via the introduction of a short deletion (tauΔex1). The tauΔex1 line was generated on a pure C57Bl/6J background and reproduced resistance to excitotoxicity in the absence of memory deficits in young mice, reported for earlier tau–/– lines [7, 21].
MATERIALS AND METHODS
Animals
Generation of tauΔex1 mice
The strategy for successful disruption of the Mapt gene using multiplex CRISPR editing has previously been reported [29]. Briefly, two guides encompassing the start codon of the MAPT gene were selected (http://crispr.mit.edu) to excise a small (≈ 150 bp) genomic fragment, allowing for easy genotyping. Single-guide RNAs (sgRNAs) were synthesized by T7-conjugated PCR, followed by in vitro transcription [30]. Cas9 protein (50 ng/μl) and sgRNAs (12.5 ng/μl each) were subsequently diluted in microinjection buffer (8 mM Tris-HCl and 0.15 mM EDTA) and microinjected into the cytoplasm of fertilized C57Bl/6J oocytes.
Mice
TaueGFP mice have been described previously [31]. Mice of both genders were used throughout the study. Mice were housed in groups of 3 to 6 littermates in individually ventilated cages (Allentown, USA) in a special pathogen-free facility. The facility operated on a 12-h light/dark cycle (6am–6pm). Anderson’s 1/8th inch corn cob bedding (Tecniplast, Italy) was changed weekly. Wooden chew sticks, red transparent plastic houses, and paper nesting material provided enrichment. Access to water and chow was not restricted. The standard chow contained 3.5 g/kg omega-3 fatty acid derived from fishmeal ad libitum (Mouse Breeder Diet, Gordon’s Specialty Stockfeeds, Australia).
Genotyping
Genotyping of mice was done by PCR amplification of genomic DNA obtained from tail biopsies; 666 bp (wild-type) and/or 527 bp (knockout) bands were produced by PCR (95°C for 30 s, 55°C for 30 s, 72°C for 30 s, for 35 cycles). Genotyping primers were 5’-GCAATCACCTTCCCTCCATA-3’ and 5’-ATTCAACCCCCTCGAATTTT-3’.
Sequencing
Size of the deletion in intron-1/exon1 of Mapt were determined by amplification of a 666 bp and/or 527 bp fragment. Fragments were sequenced (Macrogen, Inc) using the following primer pair: 5’-CTTCTCAGTGATATGGAACCATC-3’ and 5’-CATCCTTCTCCTGATGTCCTA-3’.
Western blotting
Western blotting was done as previously described [32]. Briefly, proteins were extracted from brains of 6 weeks old mice using RIPA buffer containing 50 mM Tris-Hcl, 150 mM NaCl, 1% NP-40, 5 mM EDTA, 0.5% sodium deoxycholate, 0.1% SDS and EDTA-free protease inhibitors (Roche). Proteins were separated by SDS-PAGE followed by electro-transfer onto 0.22μm nitrocellulose membranes. For detection, primary antibodies to tau (tau1, 1:1000, Millipore), Gapdh (1:1000, Sigma), Map1A (1:500, Sigma), Map1B (1:500, Sigma), and Map2 (1:1000, Sigma) were visualized using HRP-coupled secondary antibodies (Santa Cruz) and Luminata Crescendo Western HRP substrate (Millipore).
Induced seizures
Excitotoxic seizures were induced by intraperitoneal injection of pentylentetrazole (PTZ) at 50 mg/kg body weight [33]. Seizures were scored as previously established by us [5]; 0, no seizures; 1, immobility; 2, tail extension; 3, forelimb clonus; 4, generalized clonus; 5, bouncing seizures; 6, full body extension; 7, status epilepticus.
Memory testing
Learning was assessed by using a standard Morris water maze paradigm [34]. Mice were tested over 6 consecutive days in a 150 cm diameter round tank holding opaque water and a submerged escape platform (10×10 cm), as well as 4 different cues placed around the tank. Each mouse had to complete 4 trials of 60 s. Mice that did not find the platform after 60 s were guided to the platform, where they remained for another 60 s. All sessions were recorded, and swimming patterns and escape latency to find the platform were analyzed with the Anymaze software (Stoelting Co). Traces of swimming patterns were scored based on search strategies as described in Garthe and Kempermann [35]. Briefly, swim patterns were scored as follows: 1, thigmotaxis; 2, random swim; 3, scanning; 4, chaining; 5, directed search; 6, focal search; and 7, direct swim.
Statistical analysis
Data analysis was done with the Prism 7 software (GraphPad). Statistical tests are indicated in the figure legends. All values are presented as mean±standard error.
RESULTS AND DISCUSSION
We have previously used tau–/– mice that were generated by knocking in a GFP-encoding cDNA into exon 1 of Mapt using homologous recombination in embryonic stem cells and blastocyst injection [31]. Homozygous mice lacked tau expression, and instead expressed a fusion protein comprising the first 31 amino acids of tau followed by GFP. GFP expression limited functional studies using multiple fluorescence channels. Furthermore, previous tau–/– lines, including the GFP knockin line, were generated on mixed genetic backgrounds and required introduction of selection cassettes into the genome [13].
In the present study, we aimed at generating tau–/– mice with a small genome alteration directly on a C57Bl/6J background to study the function of tau in subsequent projects. In order to introduce a small deletion at the intron -1/exon 1 junction of Mapt by CRISPR/Cas9 genome editing we designed two RNA guides, 115 bp up- and 18 bp down-stream of the transcriptional start codon (Fig. 1A). Guides and Cas9 protein were injected into the cytoplasm of fertilized C57Bl/6 oocytes. All offspring were genotyped for the presence of a deletion in Mapt (Fig. 1B). Out of two offspring, two carried homozygous deletion. A male founder with an approximately 150 bp deletion, based on the size difference of genotyping bands, was chosen to establish a tauΔex1 line by crossing with C57Bl/6J mice. A heterozygous tauΔex1 offspring of this first generation (F1) was in turn crossed with C57Bl/6J mice, before intercrossing heterozygous tauΔex1 F2 offspring to obtain homozygous tauΔex1 mice. This crossing scheme ensured that possible differences in the initial deletion of Mapt on the two alleles were not carried through to subsequent generations. Breeding performance of tauΔex1 mice was not compromised and genetic modifications were inherited at Mendelian ratios across generations. TauΔex1 mice showed no overt phenotype, and were indistinguishable from their wild-type littermates in weight and size.
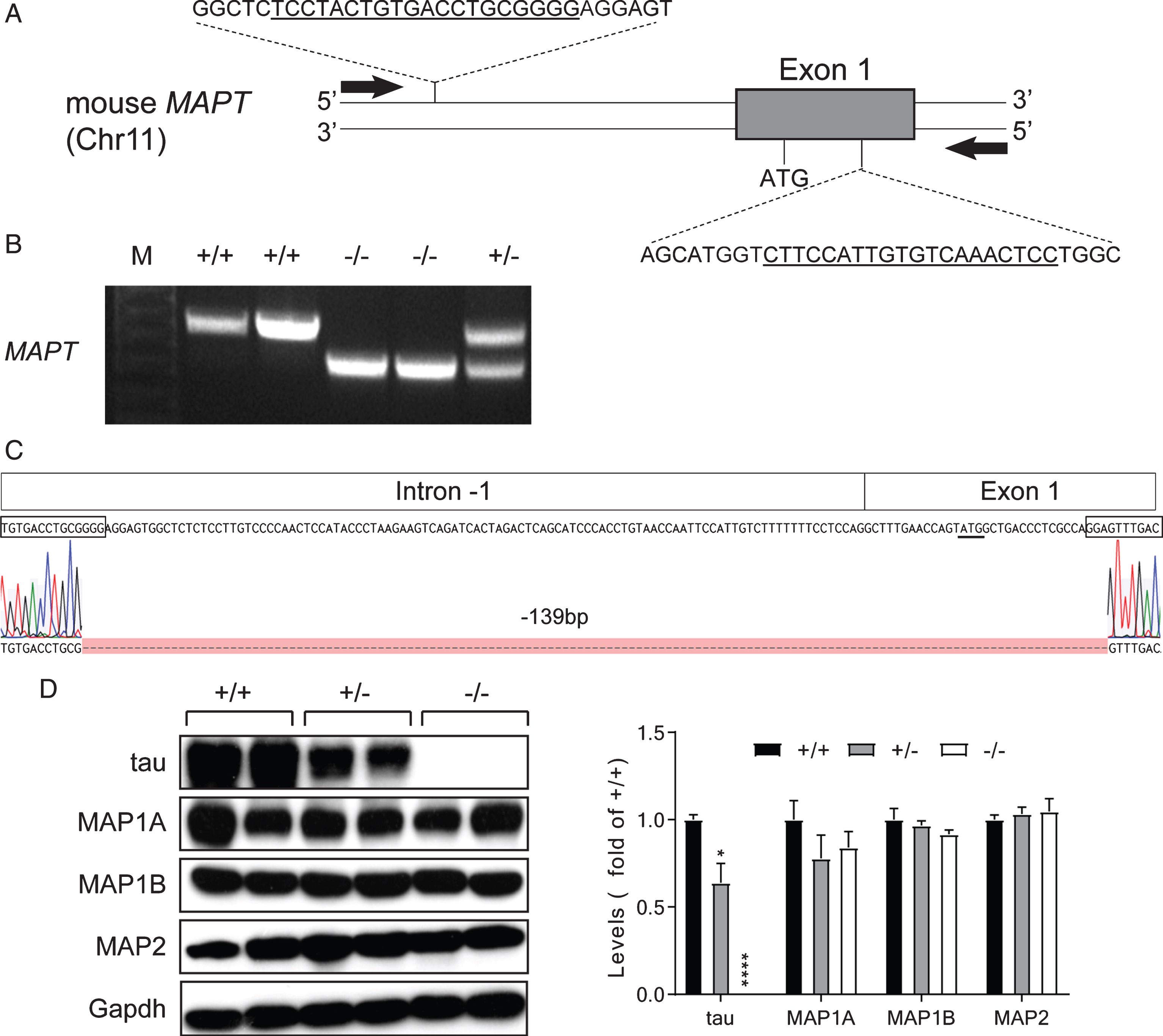
Generation of tauΔex1 mice. A) Schematic of MAPT targeting strategy. The Cas9/sgRNA-targeting sequences are underlined and the protospacer-adjacent motif (PAM) sequences are in red. Arrows indicate the position of primers used for sequencing. B) Genotyping of homozygous (–/–) and heterozygous (+/–) tauΔex1 mice and wild-type littermates (+/+); 666 bp band indicates MAPT wild-type allele and 527 bp bands the deletion allele. C) Sequencing result of the mutant MAPT allele in homozygous tauΔex1 mice confirms deletion of a 139 bp sequences including the ATG start codon (underlined), between the sgRNA sequences (boxed). D) Western blotting of brain extracts from homozygous (–/–) and heterozygous (+/–) tauΔex1 mice and wild-type littermates (+/+), showing reduction and loss of tau expression in heterozygous (+/–) and homozygous (–/–) tauΔex1 mice, respectively. Levels of MAP1A, MAP1B, and MAP2, however, were comparable in homozygous (–/–) and heterozygous (+/–) tauΔex1 mice and wild-type littermates (+/+). Quantification is shown from independent experiments (n = 6; *p < 0.05; ****p < 0.0001 [One-way ANOVA]).
To establish the exact size of the introduced deletion, we amplified a 666 bp fragment from homozygous tauΔex1 mice using primers located up- and down-stream of the predicted site in Mapt. The resulting PCR fragment was sequenced, confirming a 139 bp deletion, comprising 106 bp in the intron immediately up-stream of exon 1, plus the first 33 bp of exon 1 itself (Fig. 1C). This deletion included the transcription start codon in exon 1 of Mapt. To confirm that the deletion resulted in reduction or absence of tau protein in hetero- and homozygous tauΔex1 mice, respectively, we performed immunoblotting of brain extracts using tau-specific antibodies. This confirmed significant reduction of tau expression in heterozygous tauΔex1 mice and absence of tau protein in homozygous tauΔex1 mice (Fig. 1D). Compensatory upregulation of Map1A has been reported in one previous tau–/– strain [14]. Therefore, we also probed immunoblots for other MAP family members, including Map1A, Map1B, and Map2. We did not find changes in any of those MAP family members at 6 weeks of age (Fig. 1D). While this suggests no general compensation for the loss of tau by upregulating other MAP family members, a temporal and spatial profiling of MAP expression levels and subcellular distribution would be required to determine earlier or later changes in MAP in tauΔex1 neurons.
Others and we have previously reported that different tau–/– lines display an increased resistance to induced excitotoxic seizures [7, 21]. To confirm that this robust phenotype is also presented by our new strain, we subjected both hetero- and homozygous tauΔex1 mice and their non-mutant littermates to induced excitotoxic seizures at 6 weeks of age. Administration of PTZ (50 mg/kg i.p.) resulted in severe seizures in wild-type littermates (Fig. 2A). In contrast, the mean seizure severity was significantly reduced in heterozygous and, to a greater extend, homozygous tauΔex1 mice. Furthermore, the latency to develop more severe seizures was increased in homozygous tauΔex1 mice compared to heterozygous tauΔex1 mice and wild-type littermates for seizures of minimal severity (score 1–2) (Fig. 2B). Notably, 31% of homozygous tauΔex1 mice developed bouncing seizures (score 5) without showing prior forelimb or generalized clonus (scores 3–4) and only 6% presenting prior tail expansion (score 2) (Fig. 2C). Forty-five percent of heterozygous tauΔex1 mice developed bouncing seizures (score 5), but showed prior progression with tail extension, forelimb and generalized clonus. In comparison, bouncing seizures were observed in 80% of the wild-type littermates, with 27% and 7% reaching more severe body extension and (non-recover) status epilepticus, respectively. Taken together, tauΔex1 presented with a similar reduction in susceptibility to induced excitotoxic seizures, as did previous tau–/– strains [7, 21].
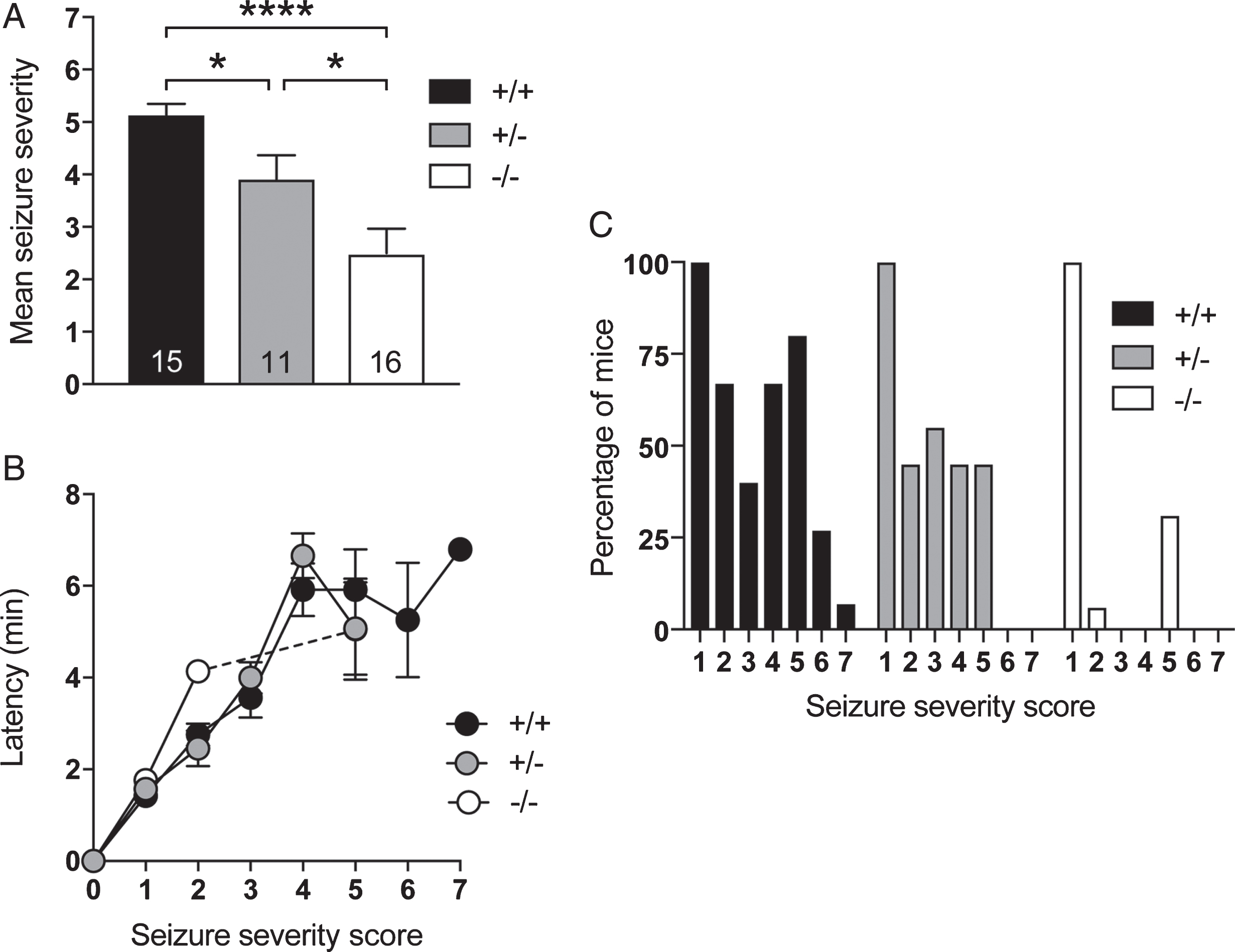
Decreased susceptibility to induced excitotoxic seizures in tauΔex1 mice. A) Mean seizure severity was significantly reduced in 6-weeks-old homozygous (–/–) and heterozygous (+/–) tauΔex1 mice compared to wild-type littermates (+/+) (n = 16 (–/–), n = 11 (+/–), n = 15 (+/+); *p < 0.05; ****p < 0.0001 [One-way ANOVA]). B) Homozygous tauΔex1 mice (–/–) showed delayed latency to develop mild seizures, while seizure progression to other stages was comparable to heterozygous (+/–) tauΔex1 mice and wild-type littermates (+/+). C) Percentage of mice reaching different seizure scores. Less heterozygous (+/–) tauΔex1 mice reached higher seizure severity scores than wild-type littermates (+/+). Only few homozygous tauΔex1 mice (–/–) progressed to even mild seizure severity, and some to seizure score 5.
Others and we have previously reported that depletion of tau has no impact on memory formation in young [6, 21] and aged [23] tau–/– mice, while again others reported deficits in aged cohorts [19]. To determine learning in tauΔex1 mice, we subjected 4-month-old animals to a standard Morris water maze paradigm and assessed the decrease in latency to find the submerged platform over subsequent testing days as an indicator of memory formation. To compare the performance of tauΔex1 to previous tau–/– mice, we included GFP knockin tau–/– (taueGFP) mice [6, 31] in these experiments. As expected, the escape latency decreased similarly over time for all genotypes, indicative of normal learning (Fig. 3A). Hence, learning was normal in young tauΔex1 mice. To determine whether there are differences in memory formation in the absence of tau, we analyzed the change in exploration strategy of individual mice over time. Following Garthe and Kempermann [35], swim traces from individual sessions were scored according to their strategy to find the submerged platform, ranging from thigmotaxis (score 1) to a direct swim path (score 7) (Fig. 3B). Irrespective of the genotype, the majority of mice showed random swim patterns upon test commencement on Day 1 (Fig. 3C). Interestingly, a larger number of homozygous taueGFP mice showed thigmotaxis on the first two testing days compared to tauΔex1 and tau+/+ mice. As testing progressed, random swimming was replaced by more successful strategies. Although by Day 5, similar proportions of homozygous tauΔex1 and taueGFP, and tau+/+ mice had developed goal-targeted swim strategies (scores 5 to 7), taueGFP maintained non-goal-targeted swim strategies (scores 3 and 4) for longer than homozygous tauΔex1 and tau+/+ mice. Taken together, absence of tau did not affect memory formation in adult mice. Future studies with aged mice are required to determine memory formation as tauΔex1 mice age.
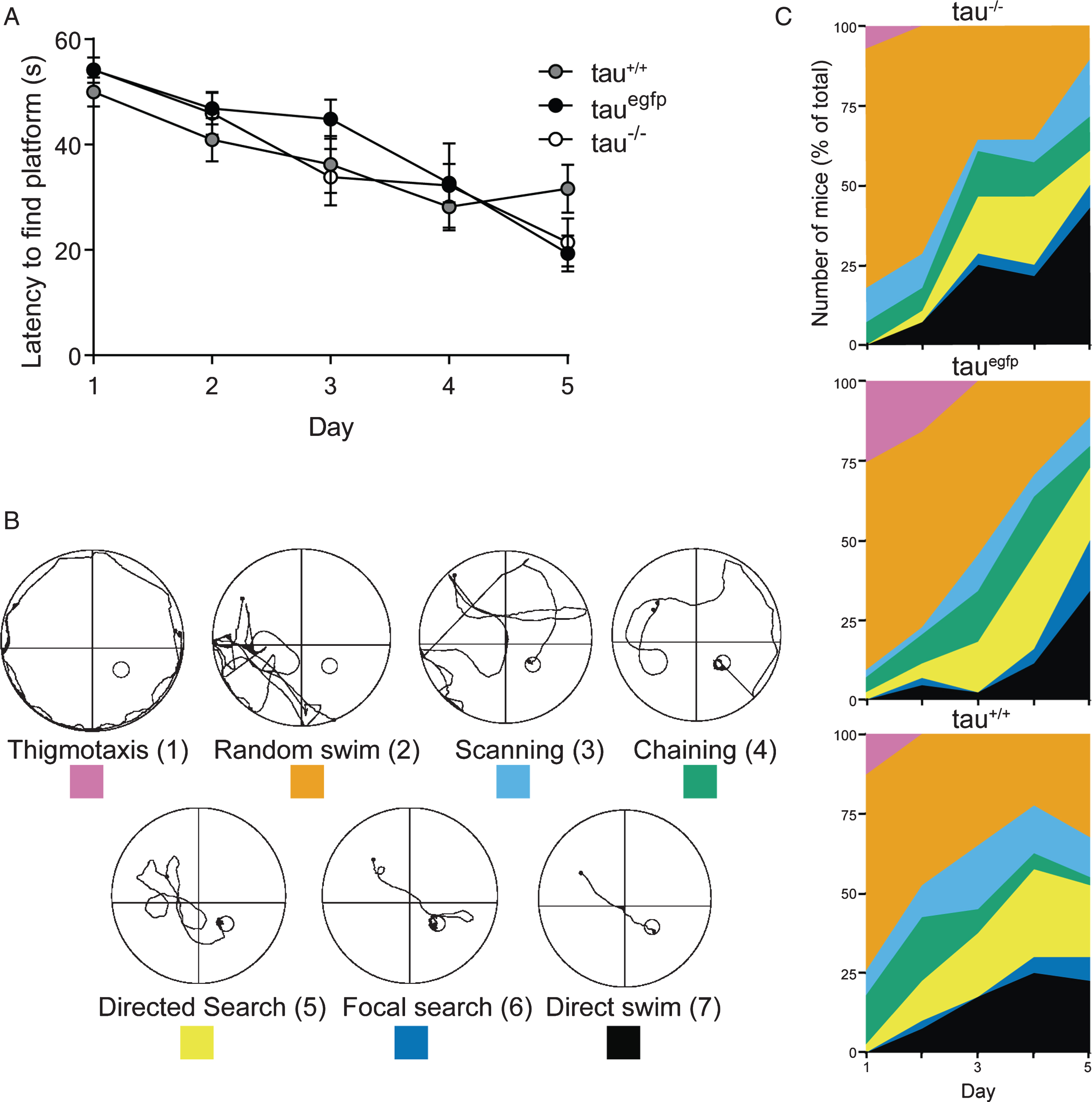
Normal memory formation in tauΔex1 mice. A) Four-month-old homozygous tauΔex1 mice (–/–; n = 7), homozygous eGFP tau knockin (tauegfp; n = 10) and wild-type (tau+/+; n = 11) mice were tested in the Morris water maze paradigm. Comparably improved latency to locate the submerged platform in all mice suggests normal memory formation. B) A schematic representation of the 7 search strategies observed in swim paths in the Morris water maze including color coding and score for each strategy. C) Prevalence of each strategy were similar in homozygous (–/–) and heterozygous (+/–) tauΔex1 mice compared to wild-type littermates (+/+), indicating comparable proficiency in spatial task.
In summary, we have generated a new tau–/– line, tauΔex1, by CRISPR/Cas9-mediated deletion of the transcriptional start in exon 1 on a pure C57Bl/6J background. TauΔex1 mice had no overt phenotypes, including normal learning in a standard test paradigm, but showed reduced susceptibility to excitotoxicity, which is in line with previous reports of other tau–/– lines [5–7, 21]. We anticipate that tauΔex1 mice will contribute to unraveling new functions of tau in physiology and disease, and may help in understanding differences between previously reported tau–/– strains. To facilitate this, we will make tauΔex1 mice available without restrictions.
Footnotes
ACKNOWLEDGMENTS
This work was supported by funding from the National Health & Medical Research Council (NHMRC) [#1037746; #1081916], the Australian Research Council (ARC) [#DP150104321; #DP170100781] and the University of New South Wales. L.I. is a NHMRC Principal Research Fellow [#1136241] and Y.K. is a NHMRC Career Development Fellow [#1123564].