
Abstract
The etiology and pathogenesis of Alzheimer’s disease (AD) are not fully understood. Thus, there are no drugs available that can provide a cure for it. We and others found that DNA polymerase-β (DNA pol-β) is required for neuronal death in several neurodegenerative models. In the present study, we tested the effect of a DNA pol-β inhibitor 2′,3′– Dideoxycytidine (DDC) in AD models both in vitro and in vivo. DDC protected primary neurons from amyloid-β (Aβ)-induced toxicity by inhibiting aberrant DNA replication mediated by DNA pol– β. Chronic oral administration of DDC alleviated Aβ deposition and memory deficits in the Tg2576 mouse model of AD. Moreover, DDC reversed synaptic loss in Tg2576 mice. These results suggest that DDC represents a novel therapeutic agent for the treatment of AD.
INTRODUCTION
Alzheimer’s disease (AD) is the most common neurodegenerative disease among old age and the most common cause of dementia. It is characterized by pathological lesions such as the deposition of neuritic plaques and neurofibrillary tangles, as well as neurodegeneration and synaptic dysfunction [1, 2]. However, the molecular mechanisms that lead to neurodegeneration and memory impairment have not been fully elucidated. Over the past decades, it has been reported that ectopic cell cycle events play causal roles in the onset and progression of AD. It is well recognized that the mature neurons in the adult brains are postmitotic cells and cannot re-enter the cell cycle. However, cell cycle regulators such as cyclins and cyclin-dependent kinases (CDKs) are found to be expressed in vulnerable neurons from AD patients [3]. Furthermore, some of the neurons have finished at least part of DNA replication. In fact, DNA replication precedes neuronal death during the onset and progression of AD [4–7]. These results indicate that the aberrant cell cycle events, especially DNA replication contribute to neuronal death in AD. Although many groups reported that inhibition of cell cycle attenuates neuronal death in some in vitro AD models, its in vivo use is halted by side effects.
We and others found that the postmitotic neurons initiate a very unique mechanism of DNA replication under stress. In postmitotic neurons challenged with amyloid-β (Aβ) or 1–methyl–4–phenylpyridinium (MPP+), DNA replication is not mediated by canonical replicative DNA polymerases such as DNA pol–ɛ or DNA pol–δ, but by DNA pol-β [8–10]. DNA pol-β is reported to be associated with base excision repair. Usually it does not mediate DNA replication in normally cycling cells [11]. Interestingly, we found that DNA replication mediated by DNA pol–β is required for neuronal death, and inhibition of DNA pol–β activity is neuroprotective [8]. Given the key roles that DNA pol–β plays in neurodegeneration, we propose that DNA pol–β inhibitors may have neuroprotective effect in AD models.
2′,3′–Dideoxycytidine (DDC) is a drug approved by Food and Drug Administration (FDA) for the treatment of human immunodeficiency virus (HIV) infection. It can specifically inhibit the activity of DNA pol–β [12, 13]. In the present study, we first examined the effect of DDC on Aβ-induced neurotoxicity in vitro. We found that DDC protected primary cortical neurons from Aβ-induced toxicity. Then we administered DDC orally to the Tg2576 mouse model of AD. Tg2576 mouse line is a transgenic AD model expressing the Swedish mutation of amyloid precursor protein (APPK670N, M671L). Aβ deposition and behavioral deficits are observed around 9–12 months of age [14]. We treated the mice with DDC or vehicle for 6 months, beginning at 6 months of age. Mice were assessed for AD-like neuropathology and cognitive performance at 12 months of age. We found that chronic oral administration of DDC prevented Aβ deposition and spatial memory deficits. DDC also reversed the loss of hippocampal synapses in this mouse model.
MATERIALS AND METHODS
Primary neuron culture
Cerebral cortical cultures were prepared from 18-day mouse embryos as previously described [15]. 10μM cytosine arabinoside was added to the medium 24 h after plating and maintained for 24 h to prevent glial proliferation. The neurons were subsequently maintained in serum-free neurobasal medium (Invitrogen, Carlsbad, CA) containing 2% B27 supplement and 2 mM glutamine at 37°C in a humidified 5% CO2 incubator. All the experiments were conducted after 10 days of in vitro culture.
Preparation of pre-formed Aβ fibrils
Hexafluoroisopropanol-treated Aβ1–42 (from rPeptide) was dissolved at 1 mg/ml in ice-cold distilled water and vortexed occasionally for 30 min. Then added 0.1 volume of 10X fibril-forming buffer (0.2 M NaPO3, 1.5 M NaCl, 0.2% NaN3, pH7.5) and vortexed. The sealed tube was place at 37°C for one week and vortexed daily. The formation of fibrils was confirmed by incubating with thioflavin S and read at excitation wavelength length of 440 nm and emission wavelength length of 485 nm.
DAN pol-β siRNA transfection
DAN pol-β siRNA and control siRNA was bought from Santa Cruz Biotechnology. The siRNA was diluted in Opti-MEM medium at a concentration of 0.2 mM, and then added to diluted lipofectamine RNAiMAX reagent (ThermoFisher Scientific, 13778) in Opti-MEM medium, and incubated for 5 min. The mixture was added to the medium and incubated for 24 h before drug treatment. The expression of DNA pol-β was assessed using western blot.
Immunohistochemistry
Immunohistochemistry was performed as previously described [16]. Briefly, tissue sections were deparaffinized and hydrated. After antigen-retrieval in boiling 10 mM sodium citrate (pH 6.0) for 20 min, the sections were incubated with an anti-Aβ antibody (Sigma) overnight at 4°C. The signal was developed using Histostain-SP kit (invitrogen). The number of plaques was analyzed using ImageJ software (National Institute of Health).
Western blot analysis
The cells were lysed in lysis buffer (50 mM Tris, pH 7.4, 40 mM NaCl, 1 mM EDTA, 0.5% Triton X–100, 1.5 mM Na3VO4, 50 mM NaF, 10 mM sodium pyrophosphate, 10 mM sodium β-glycerophosphate, supplemented with protease inhibitors cocktail), and centrifuged for 15 min at 16 000 g. The supernatant was boiled in SDS loading buffer. After SDS-PAGE, the proteins were transferred to a nitrocellulose membrane. Western blotting analysis was performed with a variety of antibodies. The anti-DNA pol–β and anti-β tubulin antibodies were purchased form Santa Cruz Biotechnology (Santa Cruz, CA). The anti-synaptotagmin antibody was purchased from Sigma-Aldrich (St. Louis, MO). The anti- spinophilin antibody was purchased from Cell Signaling Technology (Boston, MA).
Animals and treatments
Tg2576 mice were obtained from the Jackson laboratory (Bar Harbor, ME), All animal procedures were carried out in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals. 12 mice were used for each group. Mice received either DDC or vehicle orally. The treatment started at 6 months of age, and mice were subjected to Morris water maze tests at 12 months of age, and then sacrificed for other experiments. DDC was given at 50 or 100 mg/kg body weight in drinking water (220 mg/l for the 50 mg/kg group, and 440 mg/l for the 100 mg/kg group) during the whole experiment.
Electron microscopy
Synaptic density was determined by electron microscopy as described previously [17]. After deep anesthesia, mice were perfused transcardially with 2% glutaraldehyde and 3% paraformaldehyde in PBS. Hippocampal slices were postfixed in cold 1% OsO4 for 1 h. Samples were prepared and examined using standard procedures. Ultrathin sections (90 nm) were stained with uranyl acetate and lead acetate and viewed at 100 kV in a JEOL 200CX electron microscope. Synapses were identified by the presence of synaptic vesicles and postsynaptic densities.
Aβ plaque staining
Amyloid plaques were stained with Thioflavin-S as previously described [16]. Briefly, the slides were incubated in 0.25% potassium permanganate solution for 20 min, rinsed in distilled water, and then incubated in bleaching solution containing 2% oxalic acid and 1% potassium metabisulfite for 2 min. After rinsed in distilled water, the sections were transferred to blocking solution (1% sodium hydroxide and 0.9% hydrogen peroxide) for 20 min. The sections were incubated for 5 s in 0.25% acidic acid, then washed in distilled water and stained for 5 min with 0.0125% Thioflavin-S in 50% ethanol. The sections were washed with 50% ethanol and placed in distilled water. Then the sections were covered with a glass cover using mounting solution. Quantitative assessment of plaque areas was done using ImageJ software as described previously [18].
Aβ42 ELISA
The concentrations of human Aβ42 in Tg2576 mice brain were assessed with ELISA kit according to the manufacturer’s instructions (KHB3441, Invitrogen). The mice brains were homogenized in 8X mass of 5 M guanidine HCl / 50 mM Tris HCl (pH 8.0), and incubated at room temperature for 3 h. Then the samples were diluted with cold reaction buffer (phosphate buffered saline with 5% BSA and 0.03% Tween 20, supplemented with protease inhibitor cocktail), and centrifuged at 16 000 g for 20 min at 4°C. The Aβ42 concentrations in the supernatant were determined by comparison with the standard curve.
Morris Water maze
The mice were trained in a round, water-filled tub (52–inch diameter) in an environment rich with extra maze cues. An invisible escape platform was located in a fixed spatial location 1 cm below the water surface. At the beginning of each trial, the mouse was placed in the water maze with their paws touching the wall from 1 of 4 different starting positions (N, S, E, W). Each subject was given 4 trials/day for 5 consecutive days with a 15–min inter-trial interval. The maximum trial length was 60 s and if subjects did not reach the platform in the allotted time, they were manually guided to it. Upon reaching the invisible escape platform, subjects were left on it for an additional 5 s to allow for survey of the spatial cues in the environment to guide future navigation to the platform. The temperature of the water was monitored and kept between 22 and 25°C. After 5 days of task acquisition, a probe trial was presented during which time the platform was removed and the percentage of time spent in the quadrant which previously contained the escape platform during task acquisition was measured over 60 s. All trials were analyzed for latency, swim path length, and swim speed by means of MazeScan (Clever Sys, Inc.).
DNA poly-β activity assay
DNA pool-β activity was measured as described previously [19]. Poly(dA)/oligo(dT)12–18 and dTTP were used as DNA template-primer and nucleotide substrate, respectively. 16μL of DNA poly-β (0.05 units) were mixed with different concentration of DDC in 50 mmol/L Tris / HCl (pH 7.5) containing 1 mmol/l dithiothreitol, 50% glycerol, and 0.1 mmol/l EDTA, and kept on ice for 10 min. 8μL of the inhibitor-enzyme mixtures were added to 16μL of standard reaction mixtures, and incubated at 37°C for 60 min. One unit of each DNA polymerase activity was defined as the amount of enzyme that catalyzes the incorporation of 1 nmol of dTTP into synthetic template-primers (i.e., poly(dA) /oligo(dT), A/ T = 2 / 1) in 60 min at 37°C under the normal reaction conditions. The activity without DDC was considered to be 100%.
Statistical analysis
All of the data were presented as mean±SEM. Statistical analysis were performed using either Student’s t-test (two-group comparison) or one-way ANOVA followed by the LSD post-hoc multiple comparison test. The level of significance was set for p-value < 0.05.
RESULTS
DDC protects primary cortical neurons from Aβ-induced toxicity by inhibiting DNA pol-β activity
We first determined the role of DNA pol– β in neurons challenged with pro-aggregated Aβ. Primary cultured neurons were exposed to 20μM pre-aggregated Aβ for 6 h, 12 h, and 24 h, respectively. The expression of DNA pol–β was determined by western blot. We found that exposure to pro-aggregated Aβ for 12 h increased the protein level of DNA pol–β (Fig. 1A). To determine the role of DNA pol-β in Aβ-induced neuronal death, we treated primary neurons in the presence or absence of DNA pol–β inhibitor DDC or DNA pol–β siRNA. As shown in Fig. 1B, both DDC (40μM) and DNA pol–β siRNA significantly attenuated the cell viability loss, indicating DNA pol–β plays a causal role in neuronal death induced by Aβ. Interestingly, DDC did not further enhance the survival of neurons when DNA pol–β was knockdown, indicating that the effect of DDC is dependent on the expression of DNA pol–β. To verify the inhibitory effect of DDC on DNA pol–β, we incubated purified active DNA pol–β with different concentrations of DDC and tested its activity. Indeed, DDC concentration-dependently decreased the activity of DNA pol–β (Fig. 1C).
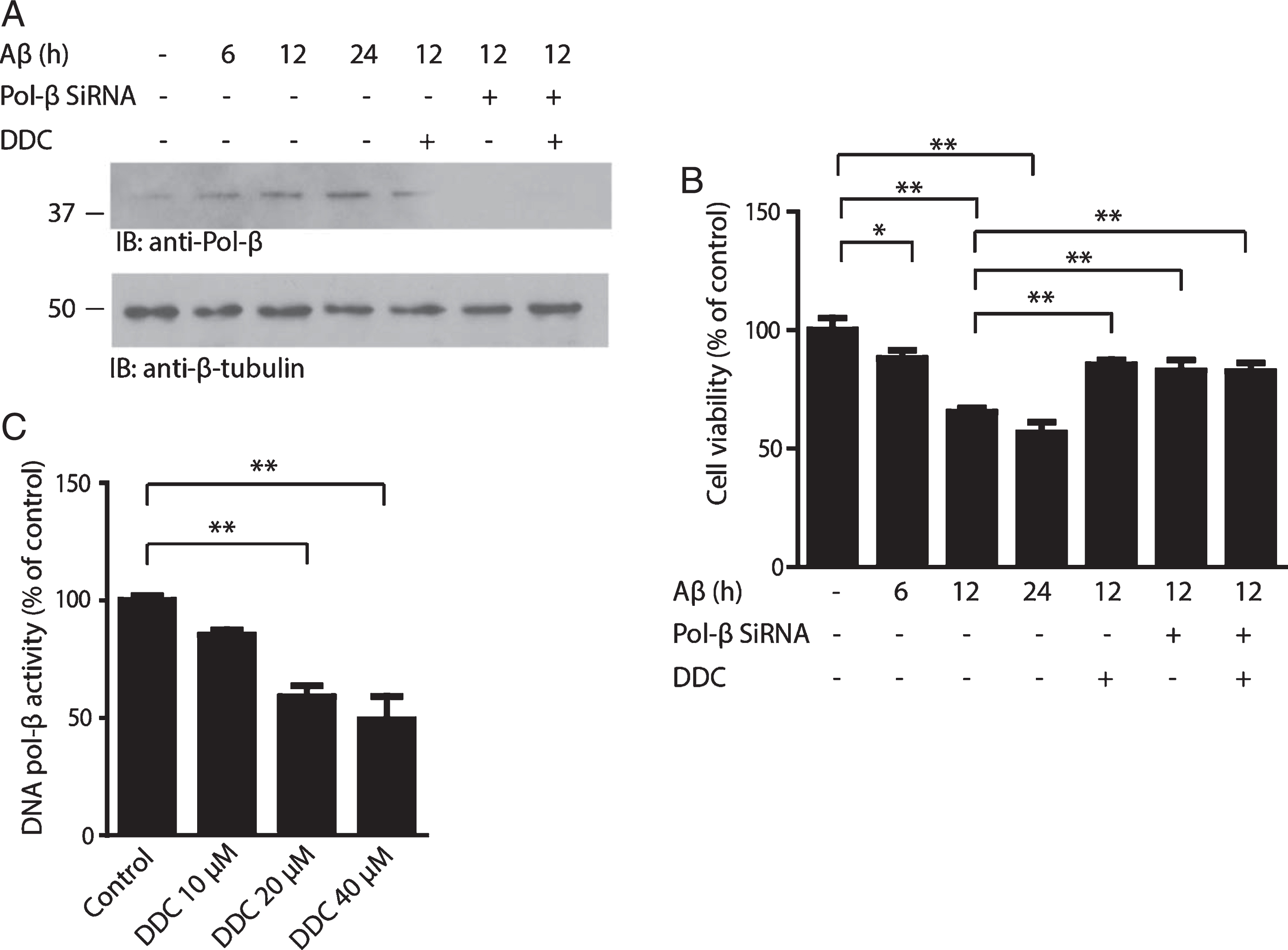
DDC attenuates Aβ-induced toxicity by inhibiting DNA pol–β activity. A) Immunoblot analysis of the expression of DNA pol–β. The neurons were incubated with 20μM pre-aggregated Aβ for 6, 12, or 24 h in the presence or absence of DNA pol–β siRNA and 40μM DDC. In the DNA pol–β siRNA group, the neurons were transfected with DNA pol–β siRNA 24 h before Aβ treatment. The protein level of DNA pol–β increased significantly after the neurons were exposed to Aβ, which was attenuated by DNA pol–β siRNA. B) Cell viability assay using MTT method. The Data are expressed as a percentage of the value in untreated control cells. C) DNA pol–β activity assay. DNA pol–β activity was determined after the purified DNA pol–β was incubated with different concentration of DDC. Date represent the mean±SEM from four independent experiments. *p < 0.05, **p < 0.01.
DDC reverses synaptic loss in Tg2576 mice
Synaptic loss is believed to be the basis of cognitive impairment in the early phase of AD [20]. Significant synaptic loss has been reported in Tg2576 mice [21]. To determine the role of DDC on synaptic dysfunction, we quantified the density of synapses in the outer molecular layer of the dentate gyrus by electron microscopy. Compared to the non-transgenic control mice, Tg2576 mice showed a significant reduction in synaptic density. DDC (50–100 mg/kg daily) treatment notably reversed the loss of synaptic density (Fig. 2A, B). We further confirmed these findings by immunoblotting using pre-synaptic markers synaptotagmin and post-synaptic markers spinophilin. Tg2576 mice showed a significant decrease in the protein level of synaptotagmin and spinophilin, indicating synaptic degeneration. DDC treatment reversed the decrease of synaptic markers (Fig. 2C). These results suggest that DDC rescues the loss of synapse in Tg2576 mice.
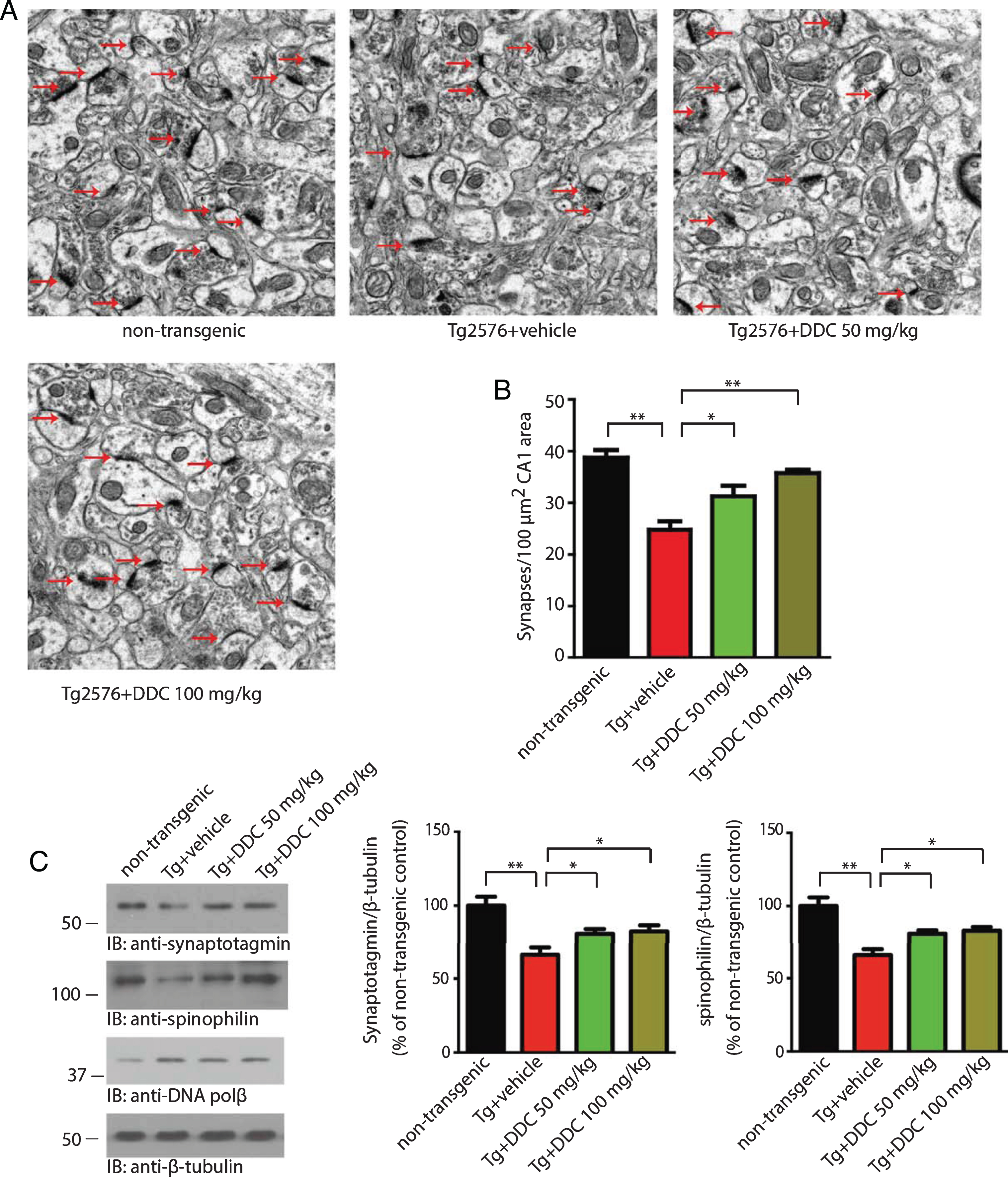
DDC reverses the synaptic loss in hippocampal CA1 area of Tg2576 mice. A) Representative electron microscopy of the synaptic structures. Arrows indicate the synapses. B) Quantitative analysis of the synaptic density in non-transgenic, vehicle- and DDC-treated Tg2676 mice. Tg2576 mice show decreased synaptic density, which was reversed by DDC. Data are shown as mean±SEM (n = 3 mice per group). *p < 0.05, **p < 0.01. C) Immunoblot analysis of synaptic markers in brain homogenates from mice treated with vehicle or DDC. The expression of presynaptic marker synaptotagmin and post-synpatic marker spinophilin were reduced in Tg2576 mice, indicating synaptic degeneration. DDC reversed the decrease of synaptic markers.
DDC alleviates Aβ deposition in Tg2576 mice
After 6 months of treatment with DDC, the deposition of Aβ was assessed by immunohistochemistry with anti-Aβ antibody. Aβ deposition was found in 12-month-old Tg2576 mice, which was alleviated by DDC treatment (Fig. 3A, B). The effect of DDC on Aβ deposition was further confirmed by Thioflavin-S staining (Fig. 3C, D). Tg2576 mice show significant plaque deposition. Strikingly, the number of plaques in the hippocampus was significantly decreased in DDC-treated mice as compared with vehicle control. We also measured the concentrations of Aβ1–42 in the Tg2576 mice brain by ELISA. The concentration of Aβ1–42 was significantly decreased by DDC treatment (Fig. 3E). These results suggest that chronic oral DDC treatment may prevent Aβ deposition and plaque formation.
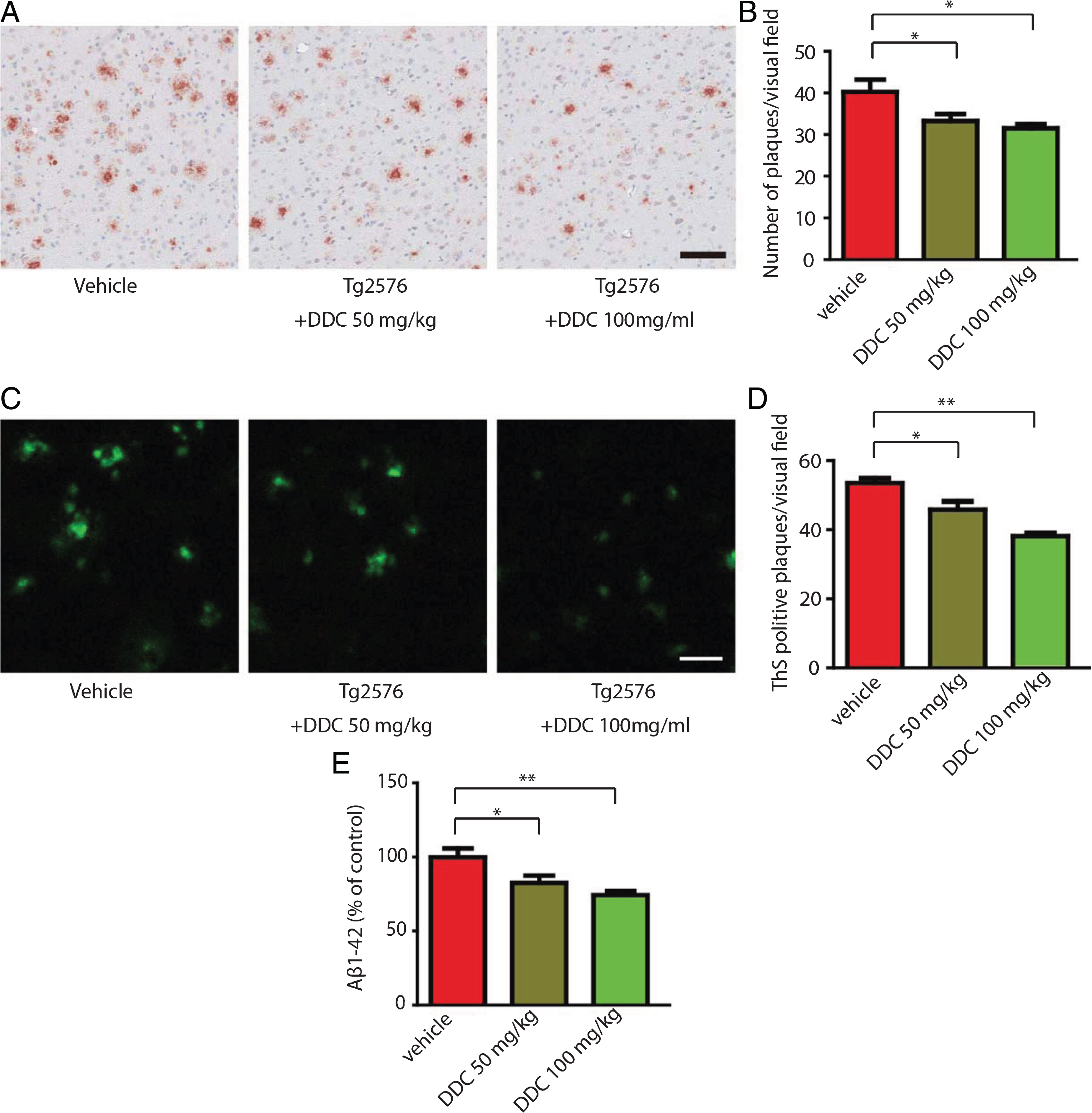
DDC alleviates Aβ deposition in Tg2576 mice. A) Immunohistochemistry of Aβ deposits in Tg2576 mice. Scale bar, 100μm. B) Quantitative analysis of amyloid plaques. C) Thioflavin-S staining of amyloid plaques in the hippocampus of Tg2576 mouse brain sections. Scale bar, 100μm. D) Quantitative analysis of the density of plaques. Amyloid deposition in Tg2576 mice was significantly decreased by DDC. n = 6 in each group. E) Aβ1–42 ELISA in vehicle- and DDC-treated Tg2576 mice. n = 6 in each group. DDC treatment decreased the concentration of Aβ1–42 in Tg2576 mice. *p < 0.05, **p < 0.01.
DDC rescues memory deficits in Tg2576 mice
Spatial memory of Tg2576 mice was tested with Morris water maze test. During the 5 acquisition days, we calculated the average latency (Fig. 4A), swim path length (Fig. 4B). Tg2576 mice showed much longer latency, longer swim path length compared to the non-transgenic control mice, indicating impaired spatial memory. DDC-treated Tg2576 mice showed shorter latency, shorter swim path length compared to water-treated Tg2576 mice. On the probe trial, water-treated Tg2576 mice spent a significantly lower percentage of time in the quadrant that formerly contained the hidden platform when compared to non-transgenic control mice. DDC treatment significantly increased the percentage of time in the target quadrant, demonstrating rescue of spatial memory recall by DDC (Fig. 4C). DDC treatment did not affect the swim speed of Tg2576 mice (Fig. 4D).
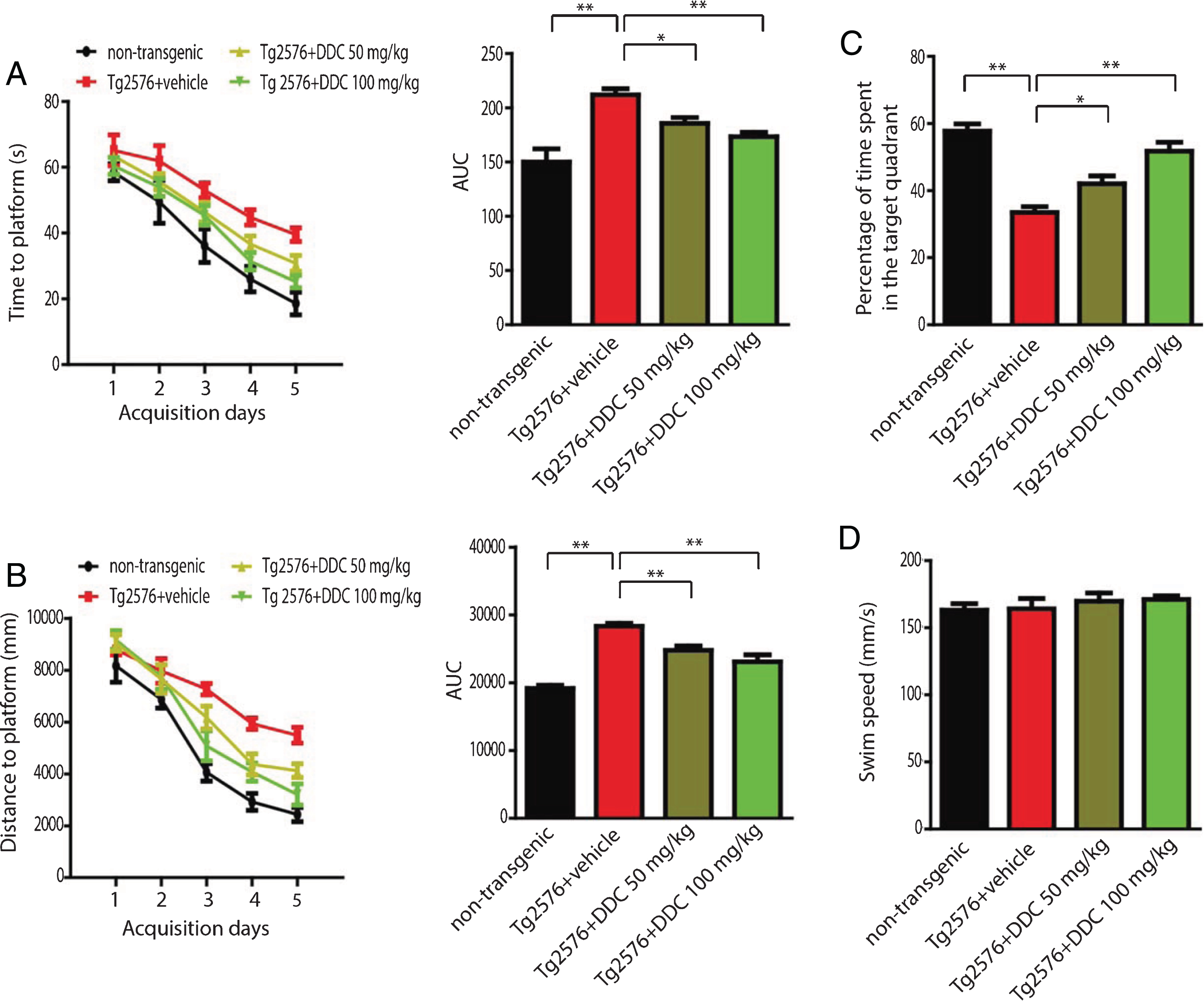
Effects of DDC on memory deficits of Tg2576 mice. Tg2576 mice and non-transgenic control mice given standard drinking water or DDC dissolved in their drinking water were trained in the water maze over five days. Shown are mean ± SEM latency to mount the escape platform (A), swim path length (B). C) A probe trial was performed on day 6 during which the platform was removed. Shown is the mean ± SEM percentage of time spent in the target quadrant. D) The swim speed of DDC- and vehicle-treated mice. *p < 0.05, **p < 0.01.
Side effects of DDC on the major organs of Tg2576 mice
To determine whether DDC exerts any toxic effects on the major organs in Tg2576 mice, we evaluated the systemic toxicity of DDC by hematoxylin and eosin staining. We did not detect any acute heart, lung, hepatic, and kidney adverse effects after the administration of DDC for 6 months (Fig. 5A). We also compared the body weight of the vehicle- and DDC-treated mice, there was no difference between the two groups (Fig. 5B).
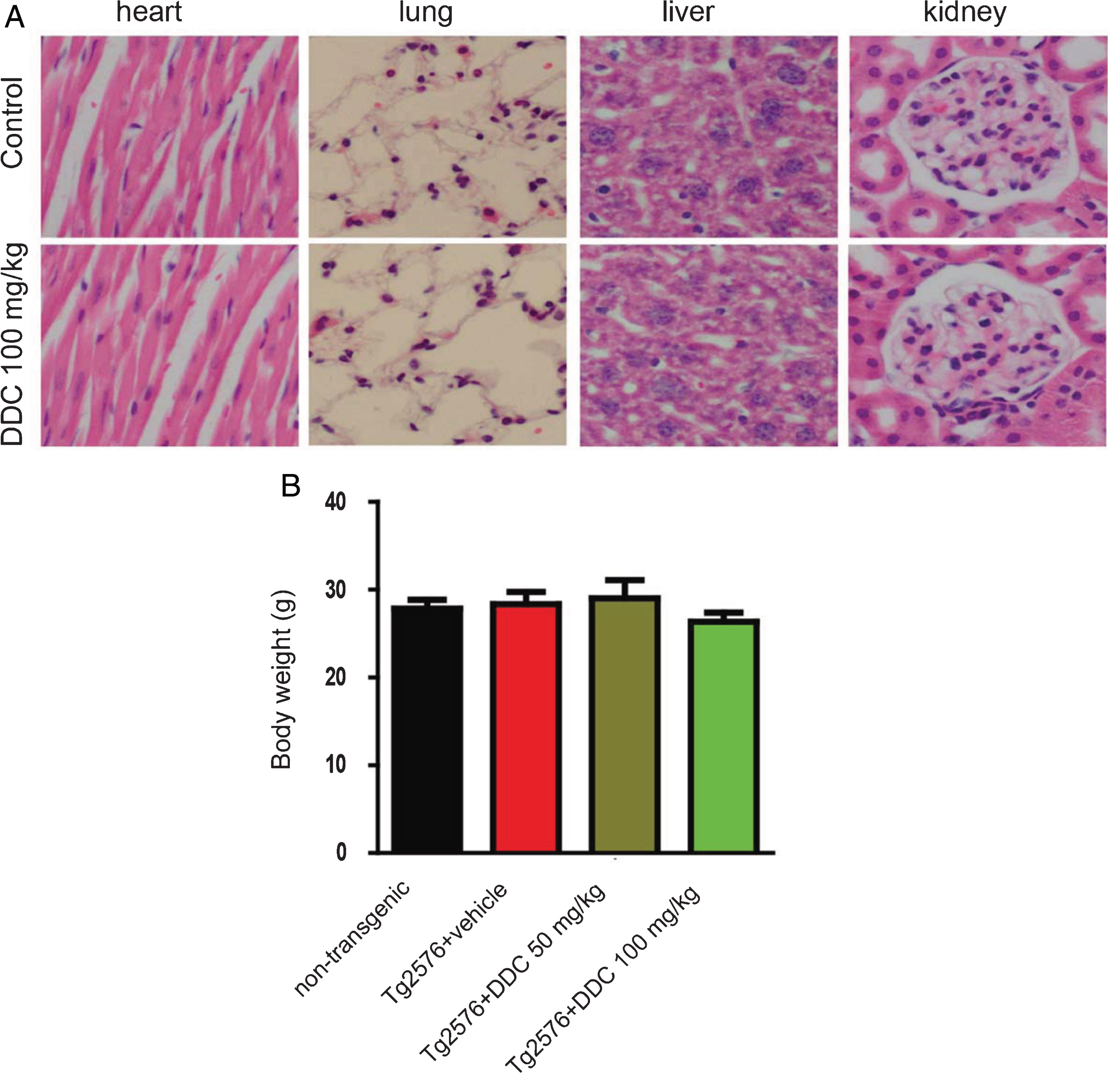
Systemic toxicity of DDC. A) Hematoxylin and eosin staining of the major organs. There is no significant toxicity on kidney, liver, lung and heart. B) The body weight of the DDC- or vehicle-treatment mice. Date represent the mean±SEM from four independent experiments.
DISCUSSION
Our results in the present study demonstrate that DNA pol–β is required for neuronal death induced by Aβ. DNA pol–β inhibitor DDC protected primary neurons form Aβ toxicity. Chronic oral administration of DDC attenuated synaptic loss, reversed Aβ deposition and rescued spatial memory deficits in a mouse model of AD.
The etiology and pathogenesis of AD has not been fully understood. However, ectopic cell cycle events have been shown to play key roles in AD pathogenesis. Although the mature neurons are exit from cell cycle, some of the degenerating neurons in AD patients aberrantly express cell cycle related factors [3]. Furthermore, there are evidence that some of the vulnerable neurons are polyploidy, indicating they have finished at least part of DNA replication [4, 22].
DNA replication is mediated by DNA polymerase. In normal cycling cells, DNA pol–δ and Pol–ɛ mediates the DNA replication. However, we and others found that a non-canonical pol–β mediates the DNA replication in AD and Parkinson’s disease models [8–10]. Furthermore, DNA pol–β is loaded into DNA replication forks in neurons challenged with Aβ [9]. In an in vitro PD model, the DNA replication mediated by DNA pol–β is required for neuronal death induced by Aβ or MPP+[8]. It is not clear how the DNA pol–β-mediated DNA replication promote neuronal death, but it has been reported that DNA pol–β is an error-prone DNA synthesizing enzyme. It replicates DNA with low fidelity compared with the “replicative” DNA polymerases (Pol–δ, Pol–α, and Pol–ɛ) [23, 24]. Conceivably, DNA replication mediated by DNA pol–β will increase the mutagenesis rate and cause genomic instability. In fact, it has been shown that DNA pol–β enhance genomic instability in cancer cells [25]. Loss of genomic stability can active cell death signals such as p53. Thus, DNA poll–β is a novel pharmacological target for the treatment of neurodegenerative diseases.
DDC is a nucleoside analog which can be metabolized to its triphosphorylated metabolite DDC-TP. DDC-TP will be incorporated into newly synthesized DNA by DNA pol–β and halt the DNA chain elongation [13]. Our in vitro data confirmed that DDC inhibited DNA pol–β activity. Furthermore, DDC protected primary neurons against Aβ toxicity. DDC did not provide further protective effect when DNA pol–β was knockdown, suggesting the effect of DDC is dependent on its inhibitory effect on DNA pol–β. DDC is an FDA-approved drug that can pass the blood-brain barrier [26–28]. We propose that oral administration of DDC may have some protective effects in AD mouse model. Lars’ group reported that subcutaneous injection of 50 mg/Kg/d results in a maximum concentration of 31.4±5.1μM DDC in the brain dialysate in rats [26]. Our group also found that an oral dose of 50 mg/Kg/d DDC is protective in a Parkinson’s disease mouse model [29]. Therefore, we treated Tg2576 mice at an oral dose of 50 mg/Kg/d.
Synaptic dysfunction occurs earlier than neurodegeneration in AD patients, and is believed to be the best pathologic correlate of dementia in AD [30, 31]. We found the synaptic density of Tg2576 mice decreased at the age of 10 month. However, DDC treatment significantly reversed the decrease of synaptic density, indicating DDC may have protective effect against the degeneration of synapse caused by the overexpression of APPswe. In agreement with this observation, the expression of synaptic markers was also increased by DDC treatment. Our results indicate that DDC is “synaptoprotective” in this AD model, which is believed to be more clinical relevance than neuroprotective therapy. The exact underlying mechanisms that mediate the protective effect of DDC on synapses remains unclear. It is possible that DDC activates the signaling pathways such as BDNF/TrkB, AKT, ERK, and exerts its “synaptoprotective” effect. Aβ oligomer is believed to play key roles in the cognitive deficits of AD. We found that the concentration of Aβ1–42 in the AD mouse brain was attenuated by DDC. Furthermore, DDC also reduced the density of plaques as demonstrated by Thioflavin-S staining and Aβ immunohistochemistry. The molecular mechanisms by which DDC attenuate the aggregation of Aβ are not clear. it is possible that DDC inhibit the activity of the amyloidogenic processing of amyloid precursor protein, or promote the degradation of Aβ. Furthermore, we did not detect any acute major side effects of DDC at the dose we used.
In summary, we found that chronic treatment of DDC has robust neuroprotective and “synaptoprotective” properties in AD models. Our study highlights DDC as a novel therapeutic strategy for the treatment of AD.