
Abstract
Currently there is no cure or effective disease-modifying therapy for Alzheimer’s disease (AD), the most common form of dementia that is becoming a global threat to public health. It is important to develop novel therapeutic strategies targeting AD pathophysiology particularly synaptic failure and cognitive impairments. Recent studies revealed several molecular signaling pathways potentially linked to brain pathology and synaptic failure in AD, including AMP-activated protein kinase (AMPK), a master kinase that plays a central role in the maintenance of cellular energy homeostasis. Particularly, hyperactive AMPK via phosphorylation has been linked to AD-associated synaptic plasticity impairments, indicating suppression of AMPK activity might be beneficial for cognitive deficiency in AD. In this review, we will discuss how targeting dysregulation of AMPK signaling could be a feasible therapeutic approach for AD.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia and one of the leading causes of disability and mortality in elderly. With the rapid growth of aging population worldwide, the prevalence and incidence of AD have been rising dramatically, becoming a global threat to public health [1]. Unfortunately, no interventions have been discovered to either slow the progress of AD or cure the disease, and recently completed clinical trials have not been successful in identifying effective disease-modifying strategies [2]. Thus, there is an urgent need to develop novel therapeutics targeting AD pathophysiology based on solid mechanistic studies. The classic brain pathological hallmarks of AD are senile plaques and neurofibrillary tangles, which consists of aggregated amyloid-β protein (Aβ) and hyperphosphorylated tau protein, respectively. However, the exact roles of Aβ and tau in AD etiology are still inconclusive. Accumulating evidence indicates failure of synaptic function as an early and key event in disease development [3 –7]. Recent studies revealed multiple molecular signaling mechanisms potentially linked to brain pathology and synaptic failure in AD, including AMP–activated protein kinase (AMPK), a sensor of cellular energy status [8]. In this review, we will discuss how targeting dysregulation of AMPK signaling could be a promising therapeutic approach for AD.
THE AMPK SIGNALING PATHWAY
AMPK was originally identified as a kinase that phosphorylated acetyl–CoA carboxylase in fatty acid synthesis and HMG–CoA reductase in cholesterol synthesis. It was later recognized that AMPK functions as a master energy sensor of cells. Essentially, AMPK is activated under low–energy conditions, leading to inhibition of anabolic reactions and promotion of catabolic processes to maintain energy homeostasis [9, 10].
Mammalian AMPKs are heterotrimeric complexes composed of a catalytic α subunit, scaffolding β and regulatory γ subunits. The two catalytic subunits of AMPK (α1 and α2) are encoded by distinct genes located on different chromosomes, and the specific targets and roles of AMPK isoforms in the neuronal system remain unclear [11 –14]. Activation of AMPK involves two molecular mechanisms in general: 1) allosteric activation by binding of AMP with the γ subunit; and 2) phosphorylation of the α subunit at Thr172 which involves either inhibition of protein phosphatases or activation of upstream kinases (Fig. 1). Phosphorylation (of Thr172) is quantitatively much more important than the allosteric activation because it causes at least 50- to 100-fold increase in AMPK activation [15]. There are three major kinases phosphorylating AMPKα at the Thr172 site including: LKB1, a tumor suppressor that appears to be constitutively active; the Ca2+ /calmodulin-dependent protein kinase CaMKKβ, which is stimulated by cytosolic Ca2+ rise; and TAK1 (also known as MAP3K7 or MEKK7), a protein kinase activated by cytokines. Notably, recent studies indicate that phosphorylation of AMPKα at sites other than Thr172 (e.g., Ser487/491) can also regulate the activity of AMPK [16 –18]. Furthermore, it has been demonstrated in non-neuronal cells that AKT is a negative regulator of AMPK, leading to increased phosphorylation at Ser485 and decreased phosphorylation of Thr172 [19, 20]. Moreover, it was reported that the detrimental effects of Aβ on synaptic plasticity are mediated by reduced AKT signaling, which presumably is a consequence of elevated caspase-3, and downstream by glycogen synthase kinase-3 (GSK3), which has been recognized as a critical player in synaptic plasticity [21] (Fig. 1). However, how AMPK is involved in the aforementioned signaling mechanisms in AD pathogenesis remains unclear.
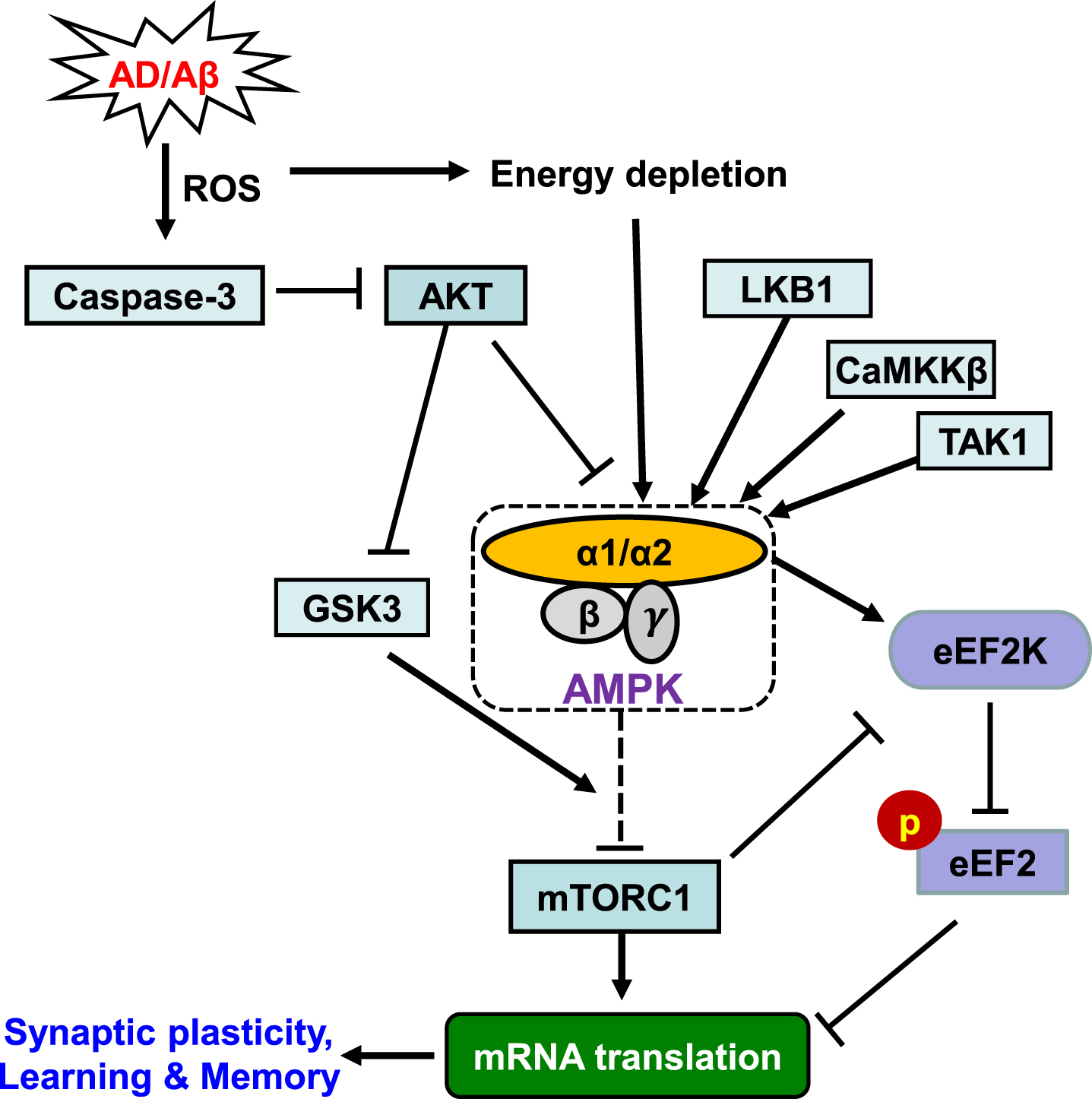
The AMPK Signaling Pathway. Schematic model depicts the main upstream regulators and downstream effectors of AMPK. Potential link to AD is indicated. Arrows denote activation and blunted lines indicate inhibition. Refer to the main text for details.
AMPK SIGNALING DYSREGULATION AND AD-ASSOCIATED SYNAPTIC FAILURE AND COGNITIVE IMPAIRMENTS
As an energy sensor regulating all aspects of cellular function, AMPK has numerous downstream targets participating in biosynthetic pathways [9]. Of particular interest for the AD field is that AMPK is capable of regulating protein synthesis (mRNA translation) and autophagy. Substantial evidence has established that de novo protein synthesis is indispensable for long-lasting forms of synaptic plasticity and memory [22 –24]. Synaptic plasticity is widely accepted as a cellular model for learning and memory, and AD is considered as a disease of “synaptic failure” [5, 25]. AMPK inhibits mRNA translation indirectly by at least two mechanisms: 1) inhibition of the mammalian target of rapamycin complex 1 (mTORC1) pathway (via phosphorylation on TSC2), which controls cap-dependent translation initiation as well as synthesis of the translational machinery; and 2) phosphorylation and activation of eukaryotic elongation factor 2 kinase (eEF2K), which turns off the elongation step in translation by phosphorylating eEF2 (Fig. 1) [26]. Recent studies suggest abnormally high levels of brain AMPK activity (as indicated by increased AMPKα phosphorylation at Thr172) in postmortem AD patients as well as in AD model mice [27, 28]. In concordance, multiple lines of evidence indicate impaired capacity of de novo protein synthesis in neurodegenerative diseases including AD and frontotemporal dementia [29 –31]. AMPK over-activation is associated with impairments of learning/memory and synaptic plasticity, and conversely, inhibition of AMPK activity using a selective antagonist compound C boosts hippocampal synaptic plasticity [27 , 33]. Notably, AD-associated impairments of long-term potentiation (LTP) and long-term depression (LTD), two major forms of synaptic plasticity and established cellular model for memory and cognition, are rescued by inhibition of AMPK activity pharmacologically [27]. Accumulating evidence implicates synaptic failure as a key event in AD pathophysiology and thus recovery of synaptic function as a potential therapeutic strategy [3], thus repression and normalization of AMPK hyperactivity might be beneficial for AD-associated cognitive impairments. Future studies are warranted to determine the roles of abnormal AMPK activity, including potential dysregulation of its different subunits and isoforms, in AD-associated cognitive impairments.
AMPK IN AD BRAIN PATHOLOGY: Aβ BIOLOGY AND TAU PHOSPHORYLATION
The neuropathological hallmarks of AD brain include extracellular plaques, mainly composed of Aβ that derives from the amyloid-β protein precursor (AβPP); and neurofibrillary tangles, mostly aggregates of hyperphosphorylated and misfolded tau protein [34]. In both pathological processes, AMPK and its associated downstream effectors have been implicated. AMPK can phosphorylate tau protein directly at multiple sites in the microtubule-binding domain and within the flanking regions [28, 35]. Hyperphosphorylation can induce self-assembly of tau proteins into paired helical filaments and subsequently to neurofibrillary tangles [36, 37]. Additionally, abnormally increased phospho-AMPK (indicating AMPK activation) was identified in the cytoplasm of cortical neurons in multiple tauopathies including AD and co-localized with phospho-tau and tangle-like structures [28]. Moreover, elevated levels of histone deacetylase 2 (HDAC2), a key player in epigenetic regulation of gene expression, has been linked to AD. A recent study revealed that HDAC2-induced tau phosphorylation in AD is mediated by AMPK activation [38]. On the other hand, AMPK may also reduce levels of tau and tau phosphorylation through various downstream mechanisms. For example, AMPK can activate silent information regulator type 1 (SIRT1), a class III protein deacetylase, leading to decreased acetylation and degradation of tau protein (including phospho-tau) since acetylation of tau inhibits the ubiquitination and degradation processes of tau protein [39]. AMPK can also affect amyloidosis through a number of potential molecular signaling mechanisms. For example, activation of AMPK reduces the β-cleavage of AβPP in cultured rat cortical neurons, while knockout of AMPKα2 increases Aβ production [40]. Reduced expression of phosphatase protein 2Cα (PP2Cα) leads to activation of AMPKα1/α2, which then reduces the expression of Aβ converting enzyme 1 (BACE1), a major AβPP cleavage enzyme, and subsequently reduces the production of Aβ [41]. AMPK can also reduce Aβ production by activating autophagocytosis through the mTORC1 signaling, which can facilitate the clearance of Aβ [42]. In contrast, there are also studies showing that activation of AMPK can lead to the biogenesis of Aβ peptides. For example, metformin, which is an activator of AMPK, can increase the biogenesis of Aβ via upregulating BACE1 expression [43].
Taken together, the involvement of AMPK in AD-associated Aβ and tau pathology is supported by ample evidence. However, whether activation of AMPK activity (under different circumstances, e.g., in vivo versus in vitro) leads to alleviation or aggravation of AD pathology still remains inconclusive. Further, given the findings that brain AMPK activity (phosphorylation) is upregulated in AD patients, AD animal models, and by exogenous Aβ application as discussed in the previous section, there clearly exists a reciprocal relationship between AMPK activity and AD brain pathology, i.e., Aβ and tau. Determination of the detailed molecular mechanisms underlying such interactions during AD progression (e.g., early versus late stage of the disease) may provide insights into the disease etiology and accordingly novel therapeutics. It would also be informative to determine how AMPK signaling is regulated with normal aging.
AMPK AS A LINKAGE BETWEEN ENERGY METABOLISM DYSFUNCTION AND AD
Dysregulation of energy metabolism has been linked to many neurodegenerative diseases including AD. Recent epidemiological studies revealed that many metabolic diseases such as obesity, diabetes, hypercholesterolemia, and several cardiovascular diseases are risk factors for cognitive impairment and sporadic AD. The association has been termed metabolic-cognitive syndrome [44]. Particularly, insulin resistance (reduced responsiveness of target tissues to normal levels of inulin), a core feature of the type 2 diabetes, has been identified as a risk factor and putative mechanisms for certain types of AD [45]. Multiple lines of evidence suggest that AMPK dysregulation is involved in the insulin resistance in AD brains. For instance, insulin resistance leads to hyperphosphorylation of AMPK at the Ser485 site (not the Thr172 site) through overactivation of AKT, leading to repression of general AMPK activity and inhibition of tau dephosphorylation induced by 5-aminoimidazole-4-carboxamide 1-β-D-ribofuranoside (AICAR) [46]. Moreover, by inhibiting AMPK activity, Aβ oligomers can decrease the surface levels of glucose transporters (GLUT) in hippocampal neurons, which results in insulin resistance [47]. Conversely, insulin resistance can increase amyloidosis via mechanisms involved repression of insulin-PI3K-AKT signaling [48], which results into upregulation of GSK-3β activity [49]. Notably, investigations on potential effects of antidiabetic drugs on AD yield conflicting results. For example, metformin, a regularly prescribed antidiabetic drug that reduces insulin resistance, has been shown to mitigate AD-like pathology in cell culture, restore Aβ transport across the blood-brain barrier and memory impairment in a mouse model of type 2 diabetes [50, 51]. On the other hand, two clinical studies suggest there might be an increased risk of cognitive impairments in patients treated with metformin [52, 53]. As also mentioned above, it was reported in cellular models of AD that metformin induced accumulation of both intracellular and extracellular Aβ that was mediated by increased beta-secretase (BACE1) expression [43]. It is worth mentioning that these studies were conducted across multiple experimental models and species. While AMPK is widely conserved across species, little is known about the exact roles of AMPK isoforms and their tissue-specific or subcellular distribution in central nervous system. Elucidation of the complexity of AMPK isoforms may help understand some of the controversy surrounding the effects of metformin on AD.
CONCLUSION
As a master kinase responsible for the maintenance of cellular energy homeostasis, AMPK plays a key role in many biological processes. In fact, dysregulation of AMPK signaling may explain many, if not all, cellular deficiencies and functional impairments in AD, considering all the downstream effectors of AMPK. Therefore, targeting AMPK can be a feasible therapeutic strategy for AD. In the meantime, controversy arises regarding whether activation or repression of AMPK activity shall be beneficial for AD pathophysiology. Based on most recent evidence indicating an association between AMPK over-activation (via AMPKα phosphorylation) and AD-associated synaptic failure, which is established as an early and key pathophysiological event during the disease progression, it would be attractive to test whether repression of brain AMPK activity could improve cognitive impairments in AD. Future investigations are necessary to elucidate the relationship between AD pathology and AMPK, particularly its subunits and different isoforms.
Footnotes
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants K99/R00 AG044469, R01 AG055581, R01 AG056622 (T.M.), F31AG055264 (H.R.Z), the Alzheimer’s Association grant NIRG-15-362799 (T.M.), the BrightFocus Foundation grant A2017457S, Wake Forest Alzheimer’s Disease Research Center (ADRC, P30AG049638) pilot grant (T.M.), Wake Forest Clinical and Translational Science Institute (CTSI) pilot grant (T.M.).