
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disease prevalent in aged people, clinically characterized by progressive memory loss, behavioral and learning dysfunction, and cognitive deficits. The pathogenesis of AD is hallmarked by formation of amyloid-β peptide aggregates (Aβ) and intraneuronal neurofibrillary tangles (NFTs), which are induced by hyperphosphorylation of amyloid-β protein precursor and tau protein, respectively. The hyperphosphorylation is controlled by cyclin-dependent kinase-5 (CDK5), the aberrant activation of which is mediated by calpain (CAPN)-induced cleavage of p35 into p25. However, the regulation of CAPN in AD remains largely unknown. Here, we studied the post-transcriptional control of CAPN1 by microRNAs (miRNAs) in the setting of AD. We found that miR-124-3p, previously reported as a miRNA that was downregulated in AD, was a CAPN1-targeting miRNA that functionally inhibited the protein translation of CAPN1 in a human neural cell line, HCN-2. In vitro, transfection with miR-124-3p reduced the levels of CAPN1 protein, the cleavage of p35 into p25, and cell apoptosis dose-dependently in HCN-2 cells. Moreover, a significant inverse correlation was detected between the levels of miR-124-3p and CAPN1 in AD specimens. Furthermore, intracranial injection of adeno-associated virus expressing miR-124-3p into APP/PS1-AD mice significantly reduced Aβ deposition and significantly improved the AD-mouse behavior in the social recognition test and plus-maze discriminative avoidance task. Together, our data suggest that post-transcriptional control of calpain by miR-124-3p plays an essential role in the development of AD.
INTRODUCTION
Alzheimer’s disease (AD) is the most common chronic neurodegenerative disease in aged people, and is clinically characterized with progressive memory loss, behavioral and learning dysfunction, cognitive decline deficits, and eventually dementia and death of the patients [1–3]. The pathological feature of AD includes accumulation of extracellular senile plaques by amyloid-β peptide aggregates (Aβ), intraneuronal neurofibrillary tangles (NFTs) mainly composed of tau protein, aberrantly functioned synapses, and neuronal cell death in the brain [1–3]. The loss of neurons primary happens in the cortex and hippocampus, resulting in deficits in learning, memorization, and other mental functions [1–5].
As mentioned above, Aβ and tau protein are the primary components of the plaques and tangles, respectively. In the past, development of therapies for AD has primarily focused on Aβ [1–3]. However, the failure of clinical trials aiming to remove Aβ in treating AD has moved the attention to tau-targeting strategies, which are based on suppression of kinases or tau aggregation [6].
A number of different post-translational modifications impair the protein degradation of tau, or increase its nuclear exclusion, leading to elevation of cytosolic tau level to increase the tau–tau interaction and polymerization, resulting in augmentation of tau aggregation and formation of NFTs [6]. Among all post-translational modifications, hyperphosphorylation appears to be the most important one. Hyperphosphorylation of tau hallmarks the early development of AD, while the degree of phosphorylation is a nice reflection of the abnormal activity of both protein kinases and phosphatases [6].
NFTs are formed by hyperphosphorylated tau and neurofilament, the production of both are mediated by cyclin-dependent kinase-5 (CDK5) [7]. CDK5 is not a typical CDK that is involved in cell cycle control, but has been well-established to regulate neuronal development [8]. CDK5 is activated by its neuronal activators p35, which is predominantly expressed in the brain [9]. Specific cleaved by calcium-dependent cysteine proteases, calpain, p35 is truncated into p25 and p10. Interestingly, p25 has the same binding potential with CDK5, but much longer half-life [10–12], compared to p35, due to the loss of p10 that has the myristoylated region for membrane targeting and the degradation signal via ubiquitin-proteosome [13]. CDK5/p25 complex causes aberrant hyperphosphorylation of various substrates of Cdk5, including amyloid-β protein precursor (AβPP) and tau, resulting in formation of Aβ and NFTs, leading to neurodegenerative pathological changes in AD [7].
As the activator of p35, the major function partner for CDK5, calpain appears to play a crucial role in the pathogenesis of AD. The major calpains are calpain 1 (μ-calpain, CAPN1) and calpain 2 (m-calpain, CAPN2), also known as conventional or classical calpains [14]. Both CAPN1 and CAPN2 are highly expressed in the central neural system (CNS). While both are present in neurons and glial cells, CAPN1 is more abundant in neurons and CAPN2 is more abundant in glial cells [15]. Some previous studies have shown an increase in calpain levels in AD [16–18], but its regulation remains largely not unknown.
Here, we studied the post-transcriptional control of CAPN1 by microRNAs (miRNAs) in the setting of AD, since it is the major type of calpain expressed in the neurons. The APP/PS1 transgenic mouse is a commonly used murine model for AD, due to its sharing with similar clinical and pathological with AD patients [19–22]. We used this model to study the effects of the interference with the specific CAPN1-targeting miRNA. We found that miR-124-3p, previously reported as a miRNA that was downregulated in AD, was a CAPN1-targeting miRNA that functionally inhibited the protein translation of CAPN1 in a human neural cell line, HCN-2. In vitro, transfection with miR-124-3p reduced the levels of CAPN1 protein, the cleavage of p35 into p25, and cell apoptosis dose-dependently in HCN-2 cells. Moreover, we detected a significant inverse correlation between the levels of miR-124-3p and CAPN1 in AD specimens. Furthermore, intracranial injection of adeno-associated virus that carries miR-124-3p under a CMV promoter into AD mice significantly reduced Aβ deposition and significantly improved the AD-mouse behavior in social recognition test and plus-maze discriminative avoidance task.
MATERIALS AND METHODS
Protocol approval
All the experimental methods including animal experiments have been approved by the research committee of Shanghai Jiao Tong University. The brain specimens were analyzed from 20 non-AD controls and 24 AD individuals, after written informed consent was obtained.
Animals
All experiments were performed in strict accordance with the Care and Use of Laboratory Animal Guideline, issued by Shanghai Jiao Tong University. Transgenic APP/PS1 mice expressing the human APPswe (K595N/M596L) and presenilin 1 (PS1ΔE9) mutants were purchased from Jackson Laboratories (Bar Harbor, ME, USA). At the age of 8 months, these mice developed Aβ deposits and exhibited significant cognitive impairment, features of AD.
Cell culture, transfection, and transduction
HCN-2 is a human cortical neuron cell line purchased from ATCC (ATCC, Rockville, MD, USA). HCN-2 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) suppled with 15% fetal bovine serum (Invitrogen, Shanghai, China) in a humidified chamber with 5% CO2 at 37°C. Plasmids carrying miR-124-3p or null or antisense for miR-124-3p (as-miR-124-3p) were constructed using corresponding sequence. Sequence for miR-124-3p: 5′-CCGUAAGUGGCGCACGGAAU-3′; Sequence for as-miR-124-3p: 5′-AUUCCGUGCGCCACU UACGG-3′. For generation of adeno-associated virus (AAV), a pAAV-CMV-GFP plasmid (Clontech, Mountain View, CA, USA) was used. The backbone also carries a GFP reporter to allow visualization of the transduced cells by green fluorescence. Human embryonic kidney 293 cell line (HEK293, ATCC) was used for virus production. The pAAV-CMV-GFP plasmid, a packaging plasmid carrying the serotype 9 rep and cap genes, and a helper plasmid (Applied Viromics, LLC. Fremont, CA, USA) were triply transfected HEK293 cells by Lipofectamine 3000 reagent (Invitrogen). A CsCl density centrifugation method was used to purify virus, the titration of which was determined by a quantitative densitometric dot-blot assay. AAVs (1011 viral particles in 50μl) were intracranially injected into the bilateral hippocampi of the AD mice. In the control group, saline of the same volume was injected.
Western blot
The cells or mouse brain tissue were homogenized in protein lysis buffer to obtain protein, the concentration of which was determined using a BCA protein assay kit (Bio-rad, Beijing, China). Western blot was done as routine, using primary antibodies including rabbit anti-CAPN1, anti-35, anti-p25, and anti-α-tubulin (Cell Signaling, San Jose, CA, USA). The secondary antibody was HRP-conjugated anti-rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). The presentative blot images were randomly selected from 5 individuals. NIH ImageJ software (Bethesda, MA, USA) was used for image acquisition and densitometric analysis of the gels.
Quantitative PCR (RT-qPCR)
Total RNA was extracted using miRNeasy mini kit (Qiagen, Hilden, Germany), and then reversely transcribed to complementary DNA (cDNA) using miScript II RT Kit (Qiagen). Quantitative PCR was performed in duplicates with QuantiTect SYBR Green PCR Kit (Qiagen). All primers were purchased from Qiagen. Values of genes were determined by sequential normalization to α-tubulin and the experimental controls.
Assessment of apoptosis
The cell apoptosis was determined with TUNEL assay and flow cytometry. For TUNEL assay, cultured cells with respective treatment was stained with a TUNEL staining Kit (Roche Applied Science, Nutley, NJ, USA). Nuclei of the cells were stained with DAPI (1 lg/ml, Roche Applied Science). For flow cytometry, cells were stained with FITC-Annexin V and propidium iodide (PI) (Becton-Dickinson Biosciences, San Jose, CA, USA), and then analyzed with FACScan flow cytometer (Becton-Dickinson Biosciences).
Assessment of Aβ plaque formation
Immunostaining for Aβ was performed using anti-Aβ antibody (EMD Millipore, Shanghai, China), as described [23]. Quantification was done based on 5 slides of distance of 50μm each other.
Bioinformatics and dual luciferase-reporter assay
TargetSan was used to predict the miRNA binding targets, as described [24]. Luciferase-reporters including the wildtype and mutate 3′-UTR were constructed (Promega, Beijing, China), and used in a dual-luciferase reporter gene assay kit (Promega), according to the manufacturer’s instruction.
Social recognition test
A social recognition test (SRT) was used to assess the social recognition memory and novelty reaction in AD-mice, as described [23]. Briefly, an empty chamber was placed in the test cage with the mice to allow spontaneous exploration. The same inducer was placed inside a transparent acrylic chamber for 5 trials of 5 min each, separated by 10 min intervals. In the last trial (5th trial), a new inducer was placed in the same acrylic chamber and the time spent sniffing was quantified again. The time spent sniffing in the social interactions was scored with a stopwatch.
Plus-Maze discriminative avoidance task
A wood-made modified plus-maze was used in the plus-maze discriminative avoidance task (PM-DAT), as described [23]. In the training session, mice were placed at the center of the apparatus, and received both the illumination of the 100 W light and cold air blow when they entered the enclosed arm containing the lamp and the hair dryer. Twenty-four hours after the training, mice were placed in the same position in the same room for 3 min without these aversive stimuli when they entered the enclosed arm with presence of non-illuminated lamp and the hair dryer. The percentage of time spent in the aversive enclosed arm during training and testing was recorded respectively for assessment of learning and memory.
Statistics
GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA) was used for statistical analysis. Analysis was done by one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test upon necessity. Spearman’s Rank Correlation Coefficients was applied for calculating bivariate correlation. All values are depicted as mean±standard deviation from 5 to 10 individuals and are considered significant if p < 0.05.
RESULTS
MiR-124-3p targets CAPN1 to suppress its translation in human neural cells
By bioinformatics analysis, we found that human CAPN1 had only three targeting miRNAs (miR-124-3p, miR-137 and miR-506-3p). Among these three miRNAs, miR-124-3p has been reported to be downregulated in AD [25–27], although in these studies BACE1 has been identified as its target. Moreover, predictive binding of miR-124-3p onto 3′-UTR of CAPN1 mRNA was conserved on both human (Fig. 1A) and mouse (Fig. 1B). Thus, here we focused on miR-124-3p, since only a downregulated targeting miRNA may be responsible for the elevated levels of CAPN1 in AD. First, miR-124-3p was overexpressed by a plasmid carrying miR-124-3p or knocked down by a plasmid carrying as-miR-124-3p in a human neural cell line, HCN-2. Transfection with a null sequence was used as a control. The RT-qPCR for miR-124-3p levels was done to confirm the alteration of miR-124-3p levels in these cells (Fig. 1C). Next, an intact 3′-UTR of CAPN1 mRNA (CAPN1 3′-UTR) and an 3′-UTR of CAPN1 mRNA with a mutant at miR-124-3p-binding site (CAPN1 3′-UTR mut) were prepared. A dual luciferase reporter assay was performed using combinations of one miR-124-3p-modifying plasmid and one CAPN1 3′-UTR plasmid, showing that the specific binding of miR-124-3p to 3′-UTR of CAPN1 mRNA is functional in HCN-2 cells (Fig. 1D). In addition, the mRNA levels of CAPN1 was not altered by modification of miR-1271 levels in HN-2 cells (Fig. 1E), but the protein levels of CAPN1 significantly decreased by overexpressing miR-124-3p, and significantly increased by knocking down miR-124-3p (Fig. 1F). Together, our data suggest that MiR-124-3p targets CAPN1 to suppress its translation in human neural cells.
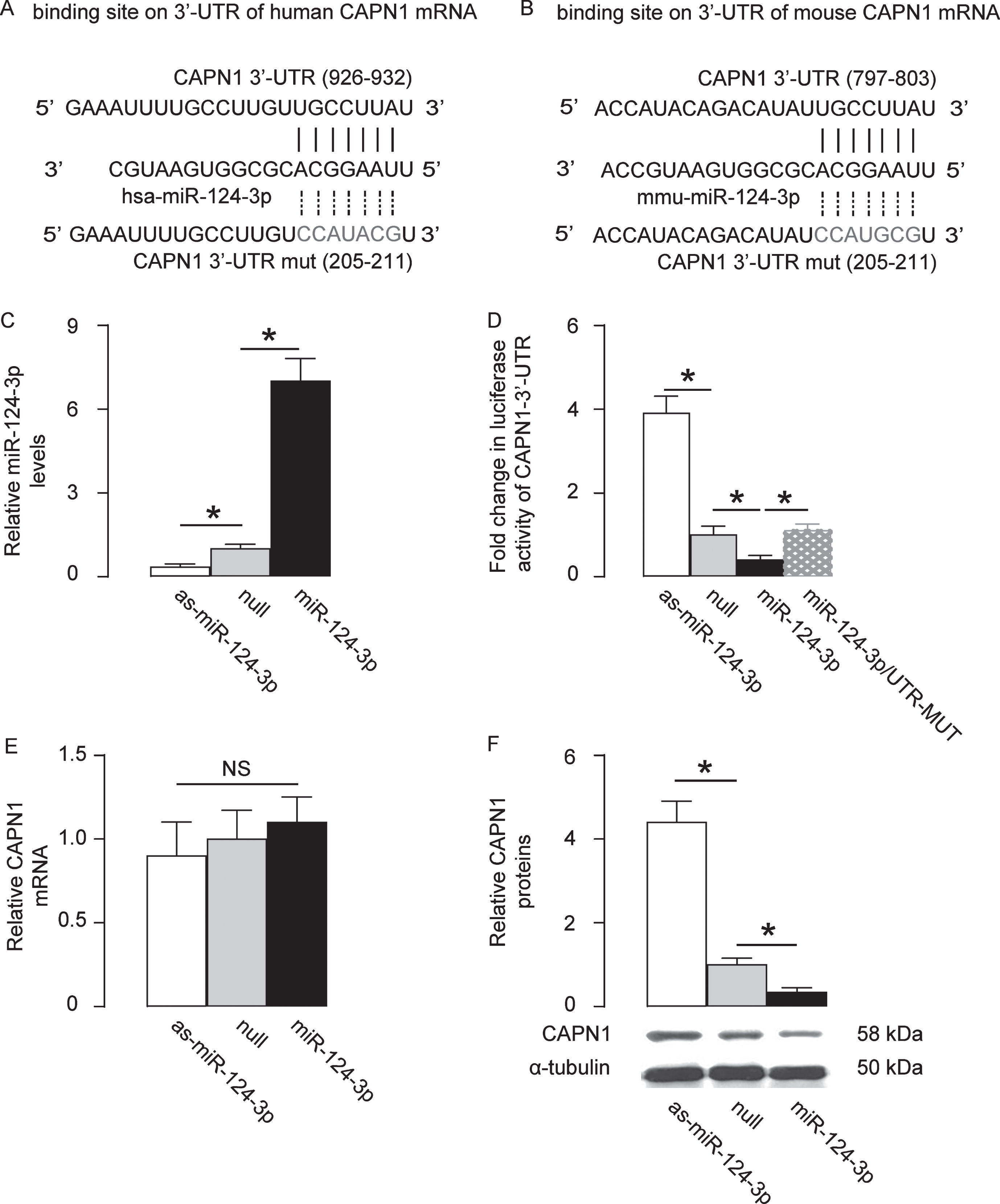
MiR-124-3p targets CAPN1 to suppress its translation in human neural cells. Bioinformatics analysis showed predictive binding of miR-124-3p onto 3′-UTR of CAPN1 mRNA in human (A) and in mouse (B). C) MiR-124-3p was overexpressed by a plasmid carrying miR-124-3p or knocked down by a plasmid carrying as-miR-124-3p in a human neural cell line, HCN-2. Transfection with a null sequence was used as a control. RT-qPCR for miR-124-3p levels in these cells. D) An intact 3′-UTR of CAPN1 mRNA (CAPN1 3′-UTR) and an 3′-UTR of CAPN1 mRNA with a mutant at miR-124-3p-binding site (CAPN1 3′-UTR mut) were prepared. A dual luciferase reporter assay was performed using combinations of one miR-124-3p-modifying plasmid and one CAPN1 3′-UTR plasmid. RT-qPCR (E) and western blot (F) for CAPN1 levels in miR-124-3p-modified HCN-2 cells. *p < 0.05. NS, non-significant. N = 5.
MiR-124-3p dose-dependently reduces the cleavage of p35 into p25 and neural cell apoptosis
Next, we examined the effects of miR-124-3p on the downstream target of CAPN1, p35. Cleavage of p35 into p25 and p10 is a key mechanism underlying hyperphosphorylation-related pathogenesis of AD. In HCN-2 cells transfected with null-plasmid or miR-124-3p, we found that miR-124-3p dose-dependently reduced the levels of CAPN1 (Fig. 2A), consistent with abovementioned results. Moreover, miR-124-3p dose-dependently reduced the cleavage of p35 into p25 (Fig. 2A), consistent with changes in the activity of CAPN1. It is well-known that cleavage of p35 and formation of p25/CDK5 induces neural cell apoptosis. Thus, we examined whether the reduction in the cleavage of p35 into p25 may affect the cell apoptosis. We found that miR-124-3p dose-dependently reduced the apoptotic cells in TUNEL assay, shown by representative images (Fig. 2B), and by quantification (Fig. 2C). Similarly, miR-124-3p dose-dependently reduced the apoptotic cells in a flow cytometry-based Annexin V assay, shown by quantification (Fig. 2D), and by representative flow charts (Fig. 2E). Together, these data suggest that miR-124-3p dose-dependently reduces the cleavage of p35 into p25 and neural cell apoptosis, the events critical for development of AD.
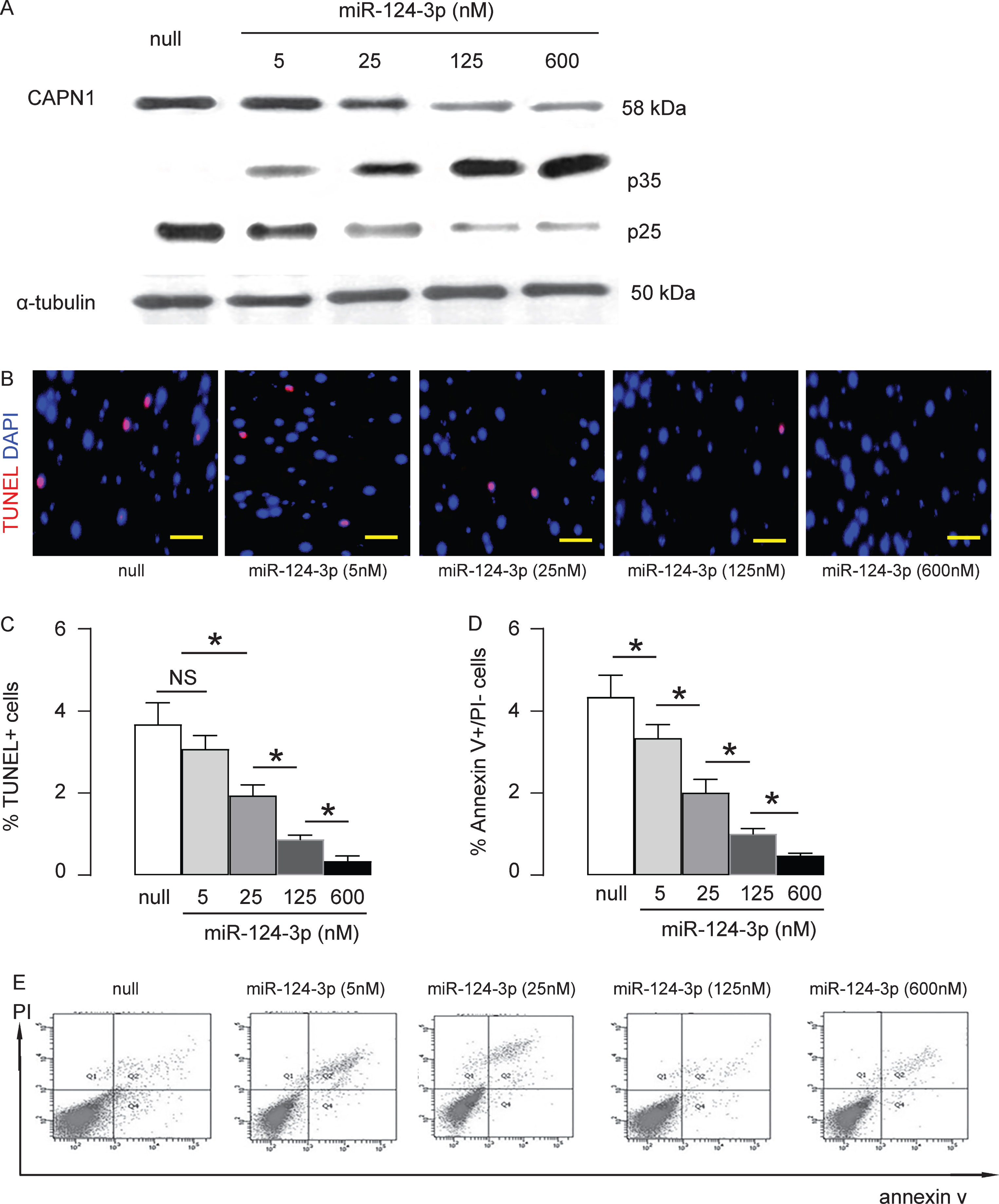
MiR-124-3p dose-dependently reduces the cleavage of p35 into p25 and neural cell apoptosis. A) Western blot for CAPN1, p35, and p25 on HCN-2 cells transfected with null, or different doses of miR-124-3p. Cell apoptosis assessed by TUNEL assay, shown by representative images (B), and by quantification (C). Cell apoptosis assessed in a flow cytometry-based Annexin V assay, shown by quantification (D), and by representative flow charts (E). *p < 0.05. NS, non-significant. N = 5. Scale bars are 20μm.
The levels of miR-124-3p inversely correlate to CAPN1 levels in AD brain specimens
In order to determine whether the miR-124-3p/CAPN1 regulatory axis has a clinical relevance, we examined the levels of them in the brain specimens from 20 non-AD and 24 AD individuals. Consistent with previous results, we found that the levels of miR-124-3p were significantly lower in AD than in non-AD (Fig. 3A), and the levels of CAPN1 were significantly higher in AD than in non-AD (Fig. 3B). The paired levels of miR-124-3p/CAPN1 were compared in the 24 AD specimens and shown a significant inverse (Fig. 3C, γ= –0.54, p < 0.001). These data suggest presence of a regulatory axis between miR-124-3p and CAPN1 in the human brain, which may contribute to the pathogenesis of AD.
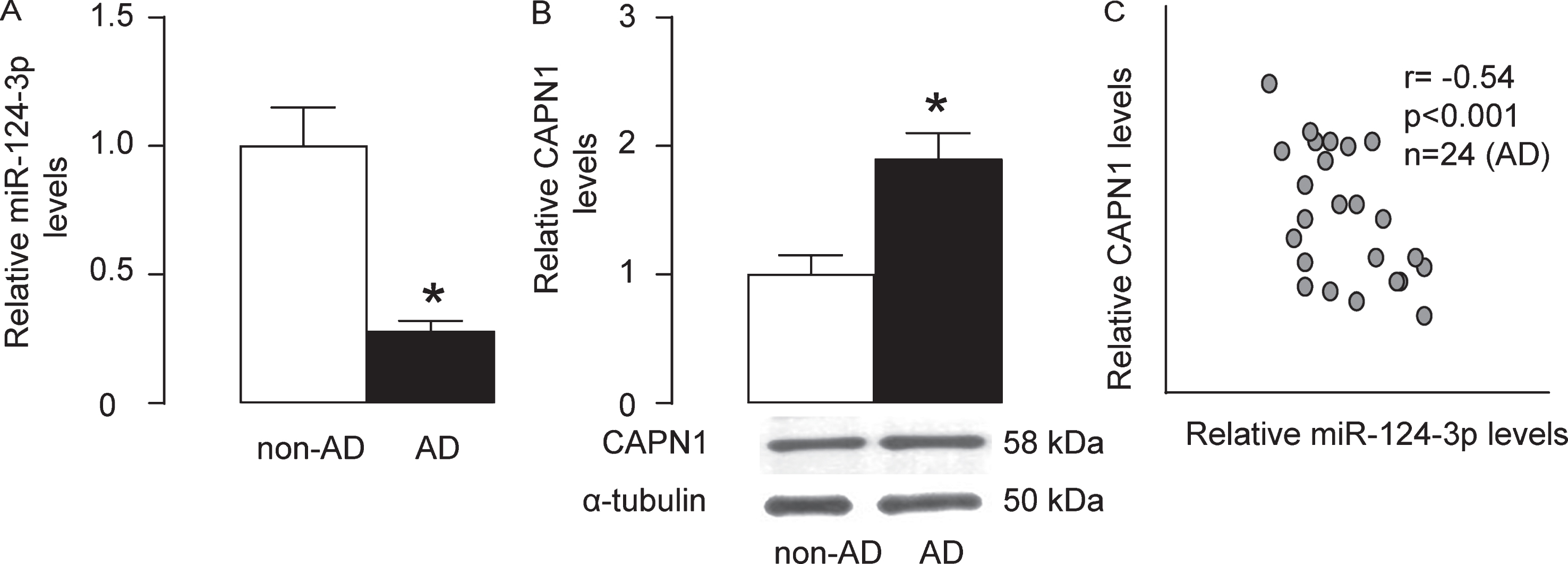
The levels of miR-124-3p inversely correlate to CAPN1 levels in AD brain specimens. The levels of miR-124-3p and CAPN1 in the brain specimens from 20 non-AD and 24 AD individuals were examined. A) RT-qPCR for the levels of miR-124-3p in AD and non-AD specimens. B) The protein levels of CAPN1 in AD and non-AD specimens. C) The paired levels of miR-124-3p/CAPN1 were compared in the 24 AD specimens and shown a significant inverse (C, γ= –0.54, p < 0.001). *p < 0.05.
Generation of AAVs that overexpress miR-124-3p
In order to assess the effects of expressing miR-124-3p in brain to compensate the loss of it on the development of AD in animal models, we prepared AAVs carrying miR-124-3p under the control of a CMV promoter. The control AAV carried null (Fig. 4A). The viral backbone had a GFP reporter, which was co-expressed with the transgene, which allowed the transduced cells to be visualized by green fluorescence (Fig. 4B). We found that the transduced cells by AAV- miR-124-3p expressed about 10 times’ higher levels of miR-124-3p than un-transduced (UnT) or null-transduced controls (Fig. 4C), resulting in about 75% knockdown of CAPN1 (Fig. 4D). Thus, these viruses were readily used for in vivo study.
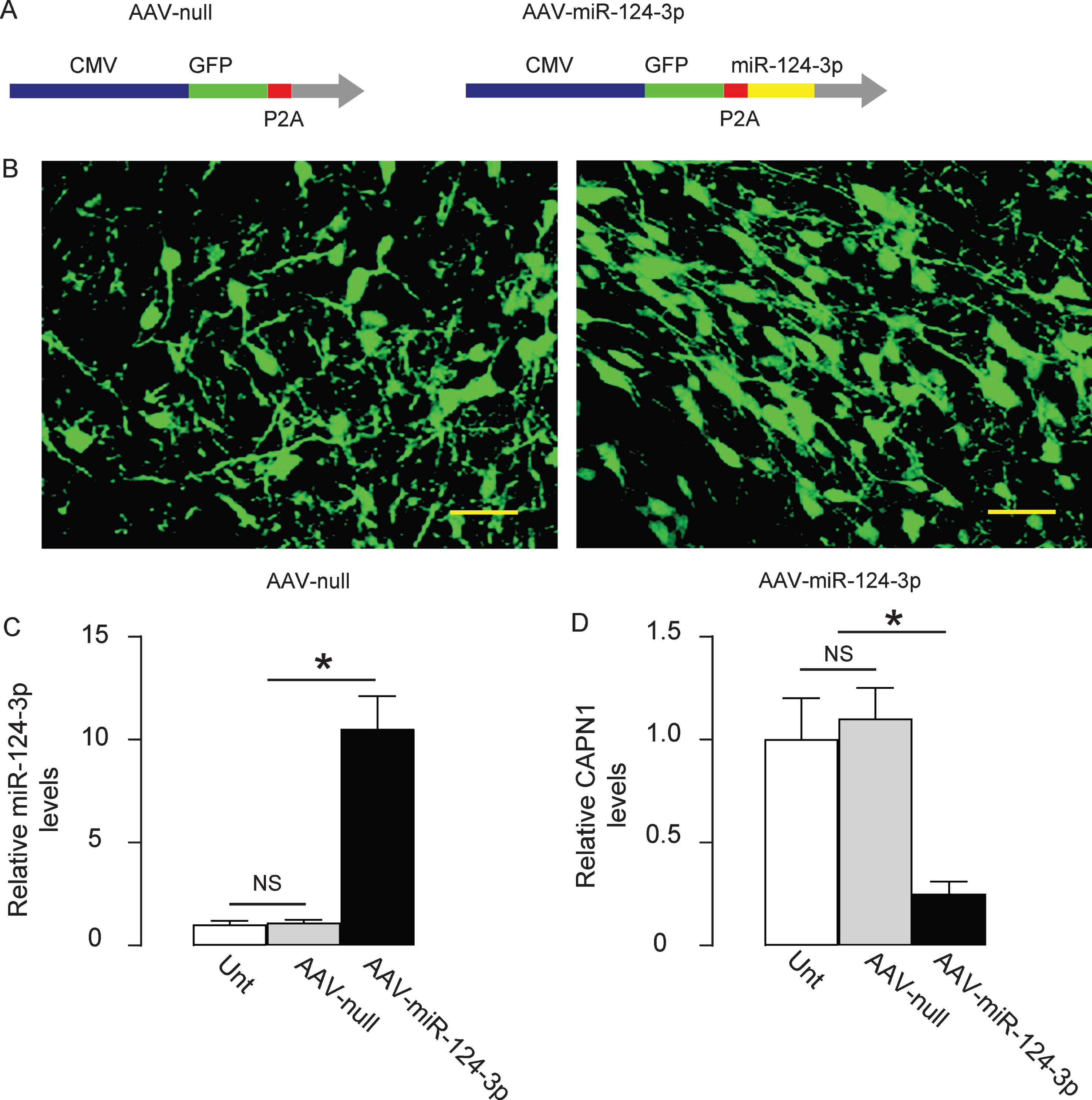
Generation of AAVs that overexpress miR-124-3p. A) Schematic showing AAVs carrying miR-124-3p under the control of a CMV promoter and AAV carrying null under the control of a CMV promoter as a control. The viral backbone had a GFP reporter, which was co-expressed with the transgene, which allowed the transduced cells to be visualized by green fluorescence. B) Transduced HCN-2 cell in culture. RT-qPCR for miR-124-3p (C) and western blot for CAPN1 (D) in AAV-miR-124-3p-transduced cells, un-transduced cells (UnT) and null-transduced cells. *p < 0.05. NS, non-significant. N = 5. Scale bars are 20μm.
Overexpression of miR-124-3p alleviates AD in mice
Then we evaluated the effects of overexpression of miR-124-3p in AD-mice. AAV-miR-124-3p or AAV-null or saline was intracranially injected into the bilateral hippocampi of the AD mice (n = 10 in each group). Ten weeks after injection, the SRT was done on these AD mice, showing significant improvement in the AD-mice that had received AAV-miR-124-3p, compared to the two control groups of AD mice (Fig. 5A). Moreover, the mice were also assessed with the PM-DAT, also showing significant improvement of the AD-mice that had received AAV-miR-124-3p, compared to the two control groups of AD mice (Fig. 5B, C). Thus, both recognition and memory of the AD mice improve by overexpression of miR-124-3p in brain. Finally, the number of Aβ plaques in the mouse brain was quantified. The senile plaques in the hippocampal stratum oriens (Oriens), lacunosum molecular (LMol), molecular (MoDG), granular (GrDG), and polymorphic (PoDG) layers were examined separately. We found that injection of AAV-miR-124-3p significantly reduced the number of Aβ plaques in any regions in mouse brain, compared to the two control groups of AD mice, shown by representative images (Fig. 5D), and by quantification (Fig. 5E). Thus, the pathological changes in the AD mice were also improved by overexpression of miR-124-3p in brain. Our results were then summarized in a schematic (Fig. 6). In a normal neuron, the presence of miR-124-3p inhibits the protein translation of CAPN1, which prevents cleavage of p35 into p25 and the subsequent formation of p25/CDK5 complex, which is responsible for hyperphosphorylation-induced AD pathogenesis. In the AD setting, loss of miR-124-3p results in elevated CAPN1, which induces cleavage of p35 into p25 and the subsequent formation of p25/CDK5 complex, which promotes the hyperphosphorylation-induced AD pathogenesis.
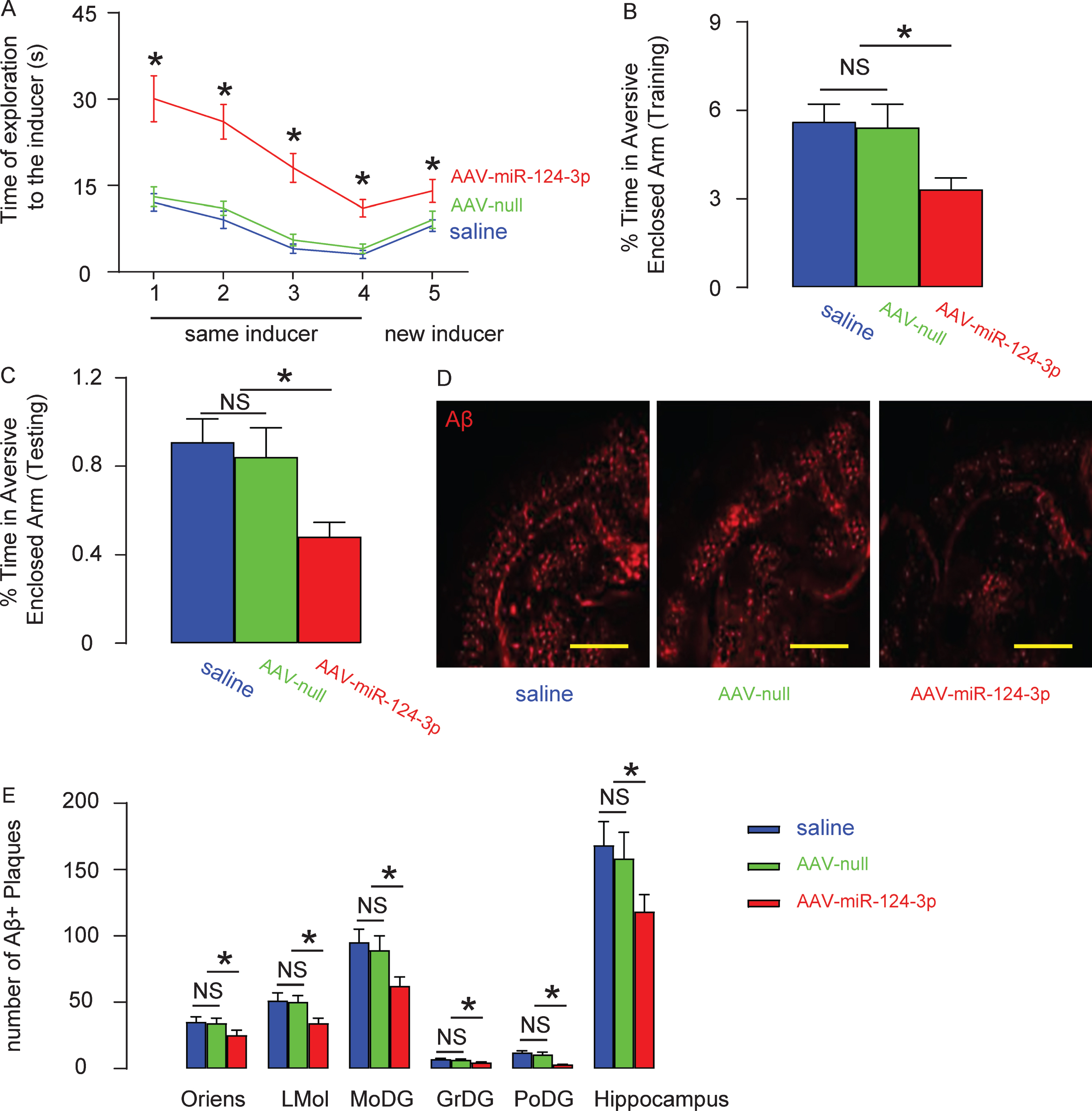
Overexpression of miR-124-3p alleviates AD in mice. AAV-miR-124-3p or AAV-null or saline was intracranially injected into the bilateral hippocampi of the AD mice (n = 10 in each group). Ten weeks after injection, mice were analyzed for Social Recognition Test (SRT; A) and Plus-Maze Discriminative avoidance Task (PM-DAT; B+C). The number of Aβ plaques in the mouse brain was quantified in hippocampal stratum oriens (Oriens), lacunosum molecular (LMol), molecular (MoDG), granular (GrDG), and polymorphic (PoDG) layers separately, shown by representative images (D), and by quantification (E). *p < 0.05. NS, non-significant. N = 10. Scale bars are 100μm.
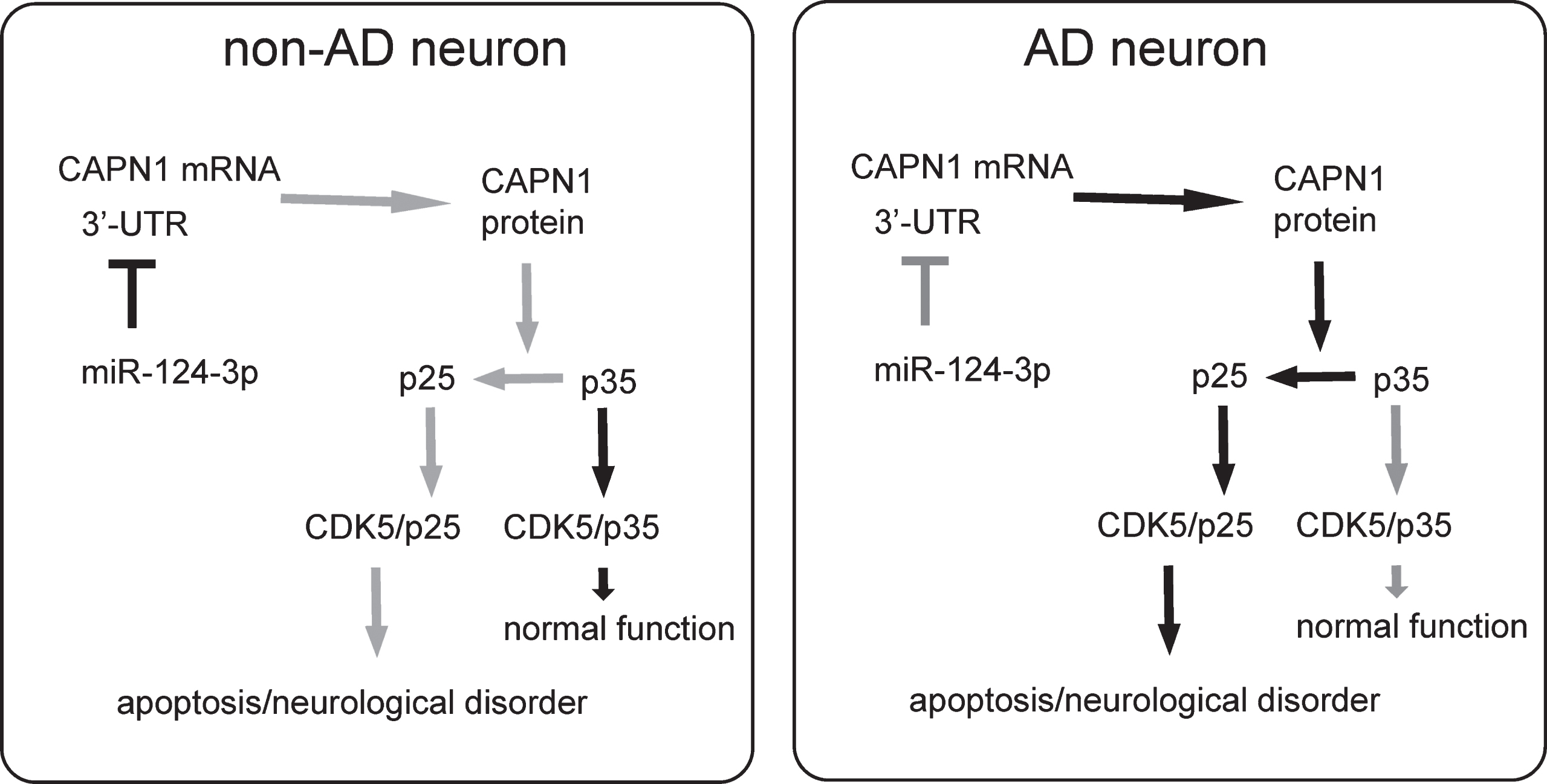
Schematic of the model. In a normal neuron, the presence of miR-124-3p inhibits the protein translation of CAPN1, which prevents cleavage of p35 into p25 and the subsequent formation of p25/CDK5 complex, which is responsible for hyperphosphorylation-induced AD pathogenesis. In the AD setting, loss of miR-124-3p results in elevated CAPN1, which induces cleavage of p35 into p25 and the subsequent formation of p25/CDK5 complex, which promotes the hyperphosphorylation-induced AD-pathogenesis.
DISCUSSION
Past studies have shown that the two major pathological changes in AD, accumulation of extracellular senile plaques by Aβ and NFTs mainly composed of tau oligomer, are both enhanced by the aberrant hyperphosphorylation of the proteins like AβPP and tau, the process primarily controlled by atypical CDK, CDK5 [7]. CDK5 can be activated by p35 or its truncated residue p25 with comparable biological function. However, the presence of degradation signal structure to be recognized by ubiquitin on p35, but not p25, makes the p25/CDK5 much stable than p35/CDK5, leading to extensive activation of CDK5 and hyperphosphorylation of AβPP and tau [13]. This well-defined molecular mechanism lacks some information on its upstream part, where the regulator for calpain, the activator for p35, is understudied.
Calpain is not only the activator of p35, and experimental suppression of calpain was shown to reduce tau phosphorylation and enhance neuronal survival in the presence of Aβ [28]. Past studies have shown that hyperactivation of calpain in the context of AD stems from enhanced intracellular Ca2+ concentration and decreased calpastatin levels [29–31], but the regulation of calpain is still poorly understood.
Here, we aimed to examine the possible regulation of CAPN1 in AD. Using TargetSan, we found that human CAPN1 had only three targeting miRNAs (miR-124-3p, miR-137, and miR-506-3p). Among these three miRNAs, miR-137 was known to play a role in the neurogenesis [32], and was downregulated in the serum from AD patients [33] and in brain samples from AD specimens [34]. The downregulation of miR-506 has also been previously noted [35]. However, the downregulation of miR-124-3p in AD has been mostly reported [25–27]. Moreover, unlike miR-137, very few functional targets of miR-124-3p have been identified. Therefore, here we focused on miR-124-3p, although we did not exclude the possibility that miR-137 and miR-506 may be interesting targets to be investigated in future. We found that the suppression of CAPN1 by miR-124-3p resulted in reduction in p35 cleavage to p25, resulting in decreased cell apoptosis as a result of reduced hyperphosphorylation by CDK5/p25, consistent with previous demonstration of the signaling cascades and their regulation of the pathological changes in AD.
We used AAVs to overexpress miR-124-3p in mouse brain, since plasmids do not allow long-term transduction of the cells, while AAVs’ mediated transduction has been shown to be persistent, induce limited inflammation, and clinically safe [36]. The social recognition paradigm is a model for assessment of social memory relying on the proper function of hippocampus. In the PM-DAT, the avoidance of the aversive enclosed arm upon testing is a measurement for retention, while the learning levels were determined based on the time spent in the aversive enclosed arm. In both assays, we found that overexpression of miR-124-3p improved the behavior of the mice. Together, the behavior and pathological scores were both improved by overexpression of miR-124-3p.
Another target of miR-124-3p is BACE1, which plays an indispensable role in the generation of Aβ [37]. Hence, the beneficial effects of miR-124-3p may be partially resulting from its suppression of BACE1, which has been shown to be a target of miR-124-3p [25–27]. Since BACE1 is also necessary for proper neural function, complete removal of BACE1 enzymatic activity may cause side effects. Thus, proper control of BACE1 may be important, which highlights miR-124-3p as a critical regulator. In the future, a systematic analysis on miR-124-3p target genes may provide a better understanding of the functional role of miR-124-3p in the pathology of AD.