
Abstract
Glycogen synthase kinase 3β (GSK3β) is a key component of pathogenesis in Alzheimer’s disease, and its inhibitors can restore cognitive function as therapeutic interventions in neurodegenerative diseases. The previous studies showed that acidic fibroblast growth factor (aFGF) could increase the phosphorylation of GSK3β through the PI3K/Akt signaling pathway. We found that aFGF14–154 markedly increased the average length of neurites in neurons damaged by amyloid-β (Aβ), and this promoting effect was blocked by GSK3β inhibitor. It is still unknown which downstream substrates of GSK3β are related to the neurite growth facilitated by aFGF14-154. The downstream substrates interacting with GSK3β were screened by co-immunoprecipitation and LTQ-Orbitrap proteomics technology in our study. Collapsin response mediator protein 2 (CRMP2) has been identified as a protein interacting with GSK3β, which is involved in the axon formation and neuron regeneration by regulating microtubule reorganization. aFGF14-154 increased the phosphorylation of GSK3β (Ser9) to inhibit its activity, then was followed by a low phosphorylation level of CRMP2 (Thr514), which led to the neurite growth. The knockdown of CRMP2 blocked the rescue of aFGF14–154 with broken neurites and shrunken cell bodies in neurons with Aβ injury. These results highlight the important role of CRMP2 and its phosphorylation through GSK3β in the effect that aFGF14-154 promoted the growth of neurite damaged by Aβ.
INTRODUCTION
Alzheimer’s disease (AD) is the main cause of progressive dementia. Senile plaques and neurofibrillary tangles, which are composed of self-polymerized amyloid-β peptide (Aβ) and hyperphosphorylated tau proteins, respectively, are the two major pathological hallmarks in the brains of patients suffering from AD [1]. Pathogenic Aβ aggregates initiate a cascade of molecular events that foster widespread neurodegeneration [2]. Excessive production and aggregation of Aβ are the initial factors and central links of AD [3]. Aβ originates from proteolysis of the amyloid-β protein precursor (AβPP) by β-secretase, the sequential enzymatic actions of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) and γ-secretase, a protein complex with presenilin 1 (PS1) at its catalytic core [4]. Abnormal hyperphosphorylation of tau protein impairs microtubule stability and cross-aggregate to form neurofibrillary tangles to damage the neurons [5].
Neurotrophic factors are important in the development, differentiation, and regeneration of neurons. Some neurotrophic factors, such as nerve growth factor and brain-derived neurotrophic factor, were explored as candidate therapeutics for AD [6, 7]. Human acidic fibroblast growth factor (haFGF) has neuroprotective effects which is similar to neurotrophic factors. aFGF is involved in the regulation of synaptic plasticity, which contributes to learning and memory ability by improving cholinergic nerve functions [8]. The level of aFGF in serum and cerebrospinal fluid is increased in patients with AD [9]. In our previous study, Liposomal Tat-haFGF14–154 (haFGF14–154 fused with Tat-PTD) treatment significantly ameliorated the behavioral deficits in APP/PS1 mice, relieved Aβ burden in the brain, and increased the expression of ADAM10 via activating the PI3K-CREB-IRE1a/XBP1 pathway [10]. The neuroprotection of aFGF against glutamate toxicity depends on GSK3β inactivation mediated by the PI3K/Akt cascades [11].
Glycogen synthase kinase 3 (GSK3) is a serine/threonine phosphokinase that widely distributed in many tissues. It has two isoforms including GSK3α (51 kDa) and GSK3β (47 kDa). The phosphorylation of GSK3 at serine site (Ser9 of GSK3β or Ser21 of GSK3α) inhibits its activity; while the phosphorylation of GSK3 at tyrosine site (Tyr216 of GSK3β, Tyr279 of GSK3α) promotes its activity [12]. The first function discovered was its role in glycogen metabolism [13]. Many studies have confirmed that GSK3β is involved in the pathological process of AD [14], including its regulation in Aβ production [15–17], the phosphorylation of tau [18, 19], and the apoptosis of neurons [20]. GSK3β affects synaptic and structural plasticity of dendritic spines [21]. Inhibiting GSK3β activity can improve learning and memory impairment [22]. GSK3β is a key component of pathogenesis in neurodegenerative diseases, and its inhibitors can restore cognitive function as therapeutic interventions [23].
Our previous studies showed that aFGF14-154 could increase the phosphorylation of GSK3β through the PI3K/Akt signaling pathway to promote neurite growth. However, it is still unknown which downstream substrate of GSK3β is involved in the neurite growth facilitated by aFGF. In this study, the downstream substrates interacting with GSK3β were screened by co-immunoprecipitation (co-IP) and LTQ-Orbitrap proteomics technology. Our results demonstrated that aFGF promoted neurite growth by regulating GSK3β-CRMP2 signaling pathway in AD models.
MATERIALS AND METHODS
Reagents
Human aFGF14–154 recombinant protein was expressed in prokaryotic expression system and purified in our laboratory as reported previously [10]. Aβ1-42 peptides were purchased from ChinaPeptides (China). Aβ1-42 dry powder was dissolved in dimethyl sulfoxide (DMSO) and diluted with sterile water. After being treated with ultrasonic sputum, Aβ1-42 was incubated in incubator for 24 h at 37°C. BIO (Selleck, USA) was dissolved in DMSO to make a stock solution. Neurobasal medium, B27 supplement and Dulbecco’s modified Eagle medium (DMEM) were purchased from Invitrogen (USA). Fetal bovine serum (FBS) was purchased from Natocor (Argentina).
Cell culture and treatments
Primary cortical neurons were separated from brains of newborn Sprague-Dawley (SD) rats (provided by the Medical Experimental Animal Center of Guangdong Province, China). Tissues were cut rapidly into small pieces in D-Hank’s buffer, and digested with 0.25% trypsin for 15 min at 37°C. Cells were cultured in Neurobasal-A Medium (Thermo Fisher Scientific, USA) at 37°C. Neurons (5 DIV) were pre-treated with aFGF14–154 for 2 h at 37°C, and 8 μM Aβ1-42 was added into the culture medium with continuous aFGF14–154 treatment for 48 h. Some cells were treated with 8 μM GSK3β inhibitor (BIO) for 30 min before aFGF14–154 treatment. N2a-APP cells (donated by Prof. Rong Liu, Huazhong University of Science and Technology) were cultured in DMEM medium (high glucose, Gibco, USA) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% FBS at 37°C.
Western blotting
Cultured cells were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, China) with PMSF (Sigma-Aldrich, USA) and Protease Inhibitor Cocktail (Roche, USA) on ice. Lysates in supernatant were separated by centrifuging at 13,000 rpm at 4°C for 30 min. The concentration of total protein was measured by BCA Protein Assay Kit (Thermo Fisher Scientific, USA). Samples were electrophoresed in 10% SDS-PAGE gel (Beyotime, China), and then electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, USA). Membranes were blocked for 1-2 h at room temperature and incubated at 4°C overnight with the following primary antibodies: rabbit anti-synaptophysin antibody (1:1000, Abcam, USA); rabbit PSD95 antibody (1:1000, Abcam, USA); mouse anti-CRMP2 monoclonal antibody (1:2000, Zen BioScience, China); CRMP2 rabbit monoclonal antibody, GSK3β rabbit monoclonal antibody, and p-GSK3β (Ser9) rabbit monoclonal antibody (1:1000, Cell Signaling Technology, USA); p-CRMP2 (Thr514) rabbit polyclonal antibody (1:1,000, affinity, USA); β-actin rabbit polyclonal antibody (1:1000, Proteintech, USA). After incubation with the secondary antibody (peroxidase-conjugated affinipure goat anti-rabbit/mouse IgG, 1:2000, Proteintech, USA) for 2 h at room temperature, the blots were detected with Super Signal West Pico chemiluminescent substrate (Pierce Biotechnology, USA).
co-IP assay and proteomics sample preparation
N2a-APP cells pellet was incubated with lysis buffer for immunoprecipitation. After debris was removed by centrifugation, the supernatant was transferred into an EP tube and immunoprecipitated with GSK3β antibody coupled with protein G Agarose beads (Sigma-Aldrich, USA) at 4°C overnight. Negative control was immunoprecipitated with IgG antibody (Santa Cruz Biotechnology, USA). Immunoprecipitants was centrifuged and washed with PBST. The protein samples were resolved by 12% SDS-PAGE and gel were stained with Coomassie Brilliant Blue R-250. The gel lane was sliced into two fractions, cut into small pieces and washed with 25 mM ammonium bicarbonate/50% acetonitrile. Then acetonitrile was added to dehydrate. Sulfhydryl bonds in the proteins within the gel were reduced by incubating with 10 mM dithiothreithol for 1 h at 56°C. Alkylation of cysteine was reduced by incubating with 55 mM iodoacetamide for 45 min. Gel pieces were rehydrated in 20 μL trypsin solution for 30 min on ice, covered with 25 mM ammonium bicarbonate and digested for 8 h at 37°C. The peptides were extracted twice with 50% acetonitrile/2% formic acid and then lyophilized to dryness.
LTQ analysis and bioinformatics analysis
The peptides were separated by a Shimadzu LC-2-AD model nanoliter liquid chromatography. The sample was reconstituted with mobile phase A (2% ACN, 0.1% FA), centrifuged (20,000 g) for 10 min, and the supernatant was aspirated. The sample flow into the trap column, then 5% mobile phase (98% CAN, 0.1% FA) for 8 min, mobile phase B (8% linear to 35%) for 35 min, mobile phase B (35% linear rise to 60%) for 5 min, mobile phase B (60% linear rise to 80%) for 2 min, 80% mobile phase B for 5 min, and 5% mobile phase B for 10 min. The sample passed nanoESI source and then entered the tandem mass spectrometer LTQ Orbitrap Velos. Other parameters set for the fragmentation are as follows: Ion source voltage at 1.6 kV, Primary mass spectrometry scanning range from 350–1600 m/z, Resolution at 70000, Secondary mass spectrometry start at 100 m/z, Resolution at 17500, Parental screening conditions for secondary fragmentation was charged 2+ to 7+, the peak intensity exceeded 10000, and the intensity was in the top 20 of the parent ion. Ion fragmentation mode was HCD. Fragment ion was detected in Orbitrap, dynamic exclusion time in 15 s. AGC set to level 3E6 and level 1E5.
To investigate the biological relevance of the identified GSK3β binding partners, bioinformatics analyses were carried out. The protein analysis through the reference sequence (RefSeq) in the NCBI system was used to classify the identified proteins into several subfamilies and families with common functions by biological process, molecular function, and cellular component. In addition, the pathway networks of the identified GSK3β binding proteins were generated by KEGG database.
Immunofluorescence
Primary cortical neurons were cultured in 15-mm circular glass coverslips at a density of 2×105 cells/well. After treatments, cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, USA) for 20 min, followed by incubation with blocking buffer (Beyotime, China) for 1 h at room temperature. Then, samples were incubated with primary antibody (mouse anti-MAP2 antibody, 1:400, Abcam, USA) overnight at 4°C. After being washed with PBS, cells were incubated with anti-mouse IgG-H&L secondary antibody (DyLight 488, 1:400, Abcam, USA) for 1 h at 37°C and kept in the dark, followed by DAPI nuclear staining (0.5 mg/mL, Beyotime) for 15 min. Images of cells were recorded by interactive laser cytometer (LSM 710 META, Carl Zeiss), and micrographs were analyzed by using Image-Pro plus 6.0 software.
Scanning electron microscopy
Cells were cultured on 15-mm circular glass coverslips in a 24-well culture plate. After treatments, samples were fixed with glutaraldehyde (Sigma-Aldrich, USA) overnight at 4°C, followed by being rinsed and dehydrated in an alcohol gradient (30%, 50%, 70%, and 90% for 3 min each and 100% for 15 min). Cells on the coverslips were incubated with isoamyl acetate (Dongzheng, China) for 5 min twice, dried by a critical point dryer, followed by gold-palladium sputtering. The coverslips were fixed on specimen mount with conductive adhesive, and the cell surfaces were examined by scanning electron microscope (Carl Zeiss, German).
siRNA transfection
The siRNA duplexes against CRMP2 were designed and synthesized by Genepharma (China) with the following sequences: GCCUCAUCAGCUAAGACAUTT (sense) and AUGUCUUAGCUGAUGAGGCTT (anti-sense). Primary cortical neurons (5 DIV) were transfected with targeted-siRNA using Lipofectamine RNAiMAX Reagent (Invitrogen, USA). The knockdown efficiency was validated by western blotting after siRNAs were transfected for 24 h.
Statistical analysis
One-way ANOVA followed by Bonferroni post hoc test was used for data analyses with statistics software (SPSS 19.0). All experimental data represent means±SEM, and p < 0.05 was considered statistically significant.
RESULTS
GSK3β is a target of aFGF ameliorating neurite damage induced by Aβ
aFGF14-154 significantly upregulated the phosphorylation level of GSK3β at Ser9 (p < 0.01 for H3000 versus Ctrl; Fig. 1A-C) and had no effect on the expression of GSK3β in N2a-APP cells. Rat primary cortical neurons (DIV5) were pre-treated with GSK3β inhibitor (1 μM BIO) for 30 min and aFGF14–154 (3000 ng/mL) for 2 h at 37°C, then 8 μM Aβ1-42 was added into the culture medium with continuous BIO and aFGF14–154 treatment for 24 h. Synaptic marker microtubule-associated protein 2 (MAP2) was stained by immunofluorescence to calculate the length of neurites.
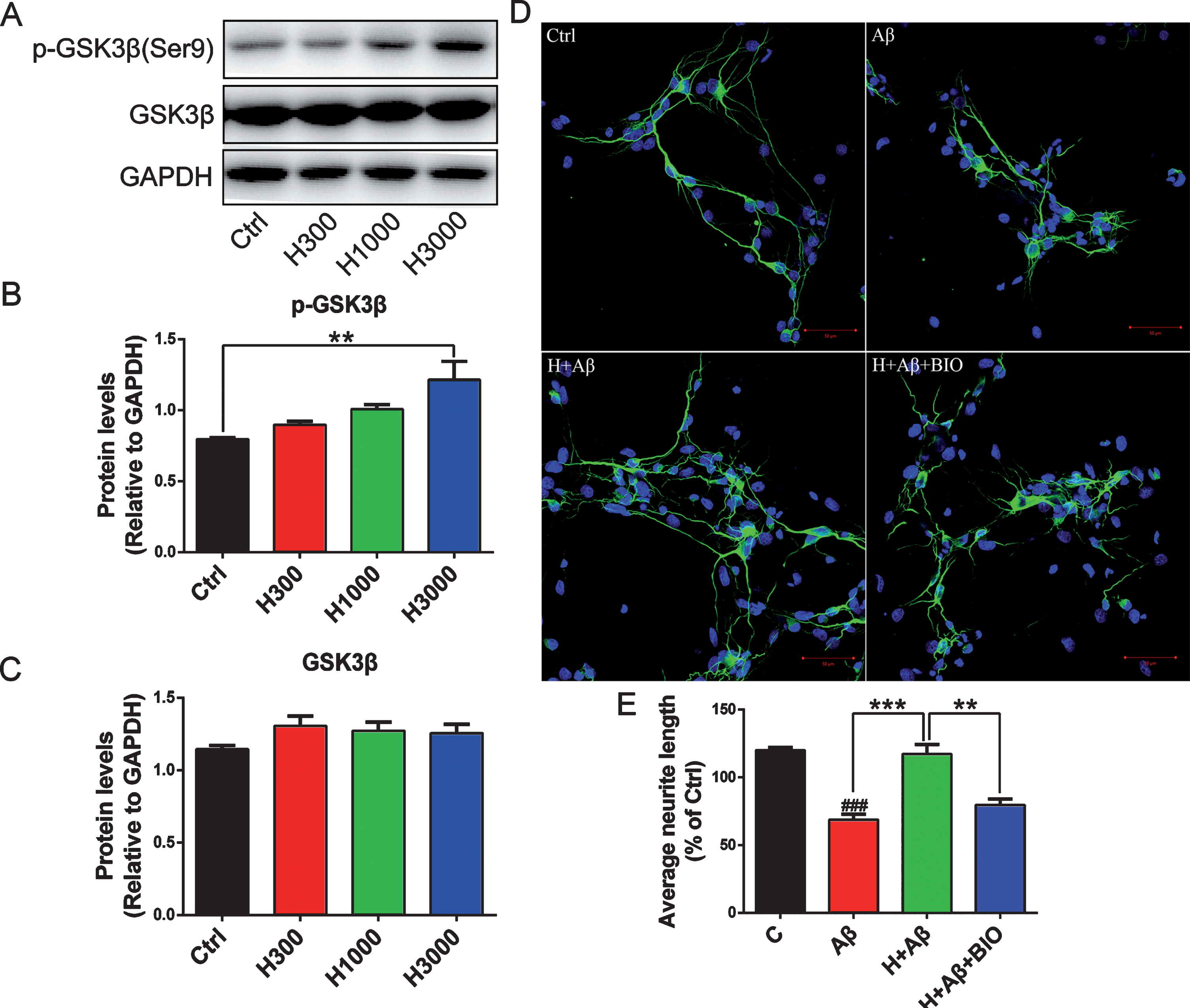
GSK3β is a target of aFGF ameliorating neurite damage induced by Aβ. Representative immunoblots (A), quantification expression of p-GSK3β (Ser9) (B) and GSK3β (C) in N2a-APP cells treated with aFGF14–154 (300 ng/mL-H300, 1000 ng/mL-H1000, 3000 ng/mL-H3000). Primary cortical neurons (DIV5) were pre-treated with GSK3 inhibitor (1 μM BIO) and aFGF14–154 (3000 ng/mL-H) for 30 min and 2 h at 37°C, respectively, then 8 μM Aβ1-42 was added into the culture medium with continuous BIO and aFGF14–154 treatment for 24 h. D) Representative immunofluorescence images of neurons with MAP2-positive staining (scale bar: 20 μm). The GSK3β inhibitor reversed the effect of aFGF14–154 on promoting neurons to regenerate. E) The quantification of neurites length. Values were presented as means±SEM (###p < 0.001 compared to Ctrl; **p < 0.01, ***p < 0.001 for significant difference, one-way ANOVA with Bonferroni post hoc test, n = 3).
Aβ1-42 damaged the neurons, and the length of neurites was significantly shorter than those in control cells (p < 0.001 for Aβ versus Ctrl; Fig. 1D, E). aFGF14–154 markedly increased the average length of neurites in neurons damaged by Aβ, and the promoting effect of aFGF14-154 was blocked by GSK3β inhibitor (p < 0.001 for H+Aβ versus Aβ, p < 0.01 for H+Aβ+BIO versus H+Aβ; Fig. 1D, E).
Identification of GSK3β interacting proteins by co-IP and LTQ proteomics technology
After N2a-APP cells were treated with aFGF14-154 for 24 h, a proteomics analysis was conducted to investigate the proteins interacting with GSK3β by co-IP. The endogenous expression of GSK3β in N2a-APP cells was confirmed by western blotting. GSK3β was detectable at the molecular weight of 46 kDa in the immunoprecipitants pulled down with anti-GSK3β antibody compared with that pulled down with anti-IgG antibody, which verified the specificity of antibody binding with GSK3β (Fig. 2A). After visualization by Coomassie brilliant blue staining, the H3000 group showed different protein bands in the molecular weight range of 130 kDa, 70–100 kDa, 55 kDa, 40–55 kDa, and 70 kDa compared with the IgG group (Fig. 2B). The proteins were followed by digestion in-gel and LTQ Orbitrap.
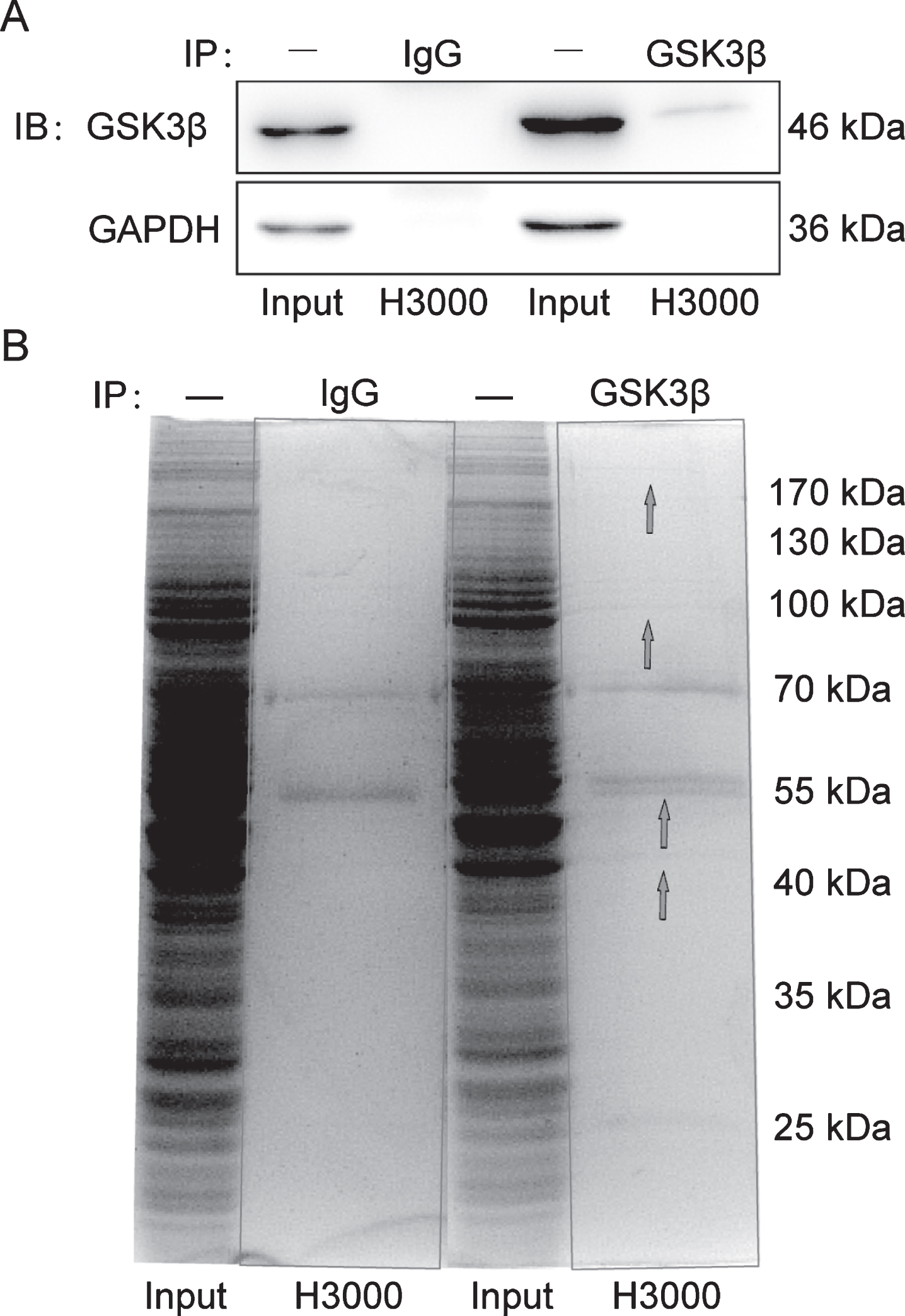
The proteins interacting with GSK3β were pulled-down by co-IP in N2a-APP cells. A) The pull-down proteins were verified by western blotting. B) GSK3β immune complexes were resolved by SDS-PAGE with Coomassie brilliant blue staining. There were several different protein bands in the molecular weight range of 40 to 170 kD (as shown by the blue arrows) in the immunoprecipitants.
Gene ontology and pathway analyses
There were 310 proteins identified as GSK3β interacting partners, and the potential functions were determined using Gene Ontology (GO) analyses. As shown in Fig. 3A, the interacting proteins were related to catalytic activity (148 proteins), structural molecule activity (33 proteins), transporter activity (20 proteins), enzyme regulator activity (15 proteins), nucleic acid binding transcription factor activity (12 proteins), protein binding transcription factor activity (12 proteins), electron carrier activity (9 proteins), receptor activity (8 proteins), molecular transducer activity (4 protein), and translation regulator activity (4 protein) in molecular function analyses. The cellular component classification results revealed that the majority of the identified GSK3β interacting proteins were involved in cell part (291 proteins), organelle (254 proteins), macromolecular complex (164 proteins), membrane enclosed lumen (117 proteins), membrane (108 proteins), extracellular region (9 proteins), synapse (9 proteins), and cell junction (7 proteins) (Fig. 3B). The analysis results of GO biological processes showed that most of the identified proteins were related to cellular process (265 proteins), metabolic process (249 proteins), single organism process (234 proteins), biological regulation (148 proteins), regulation of biological process (141 proteins), cellular component organization or biogenesis (115 proteins), response to stimulus (112 proteins), localization (109 proteins), multicellular organismal process (92 proteins), developmental process (86 proteins), positive regulation of biological process (69 proteins), signaling (67 proteins), and negative regulation of biological process (57 proteins, Fig. 3C).
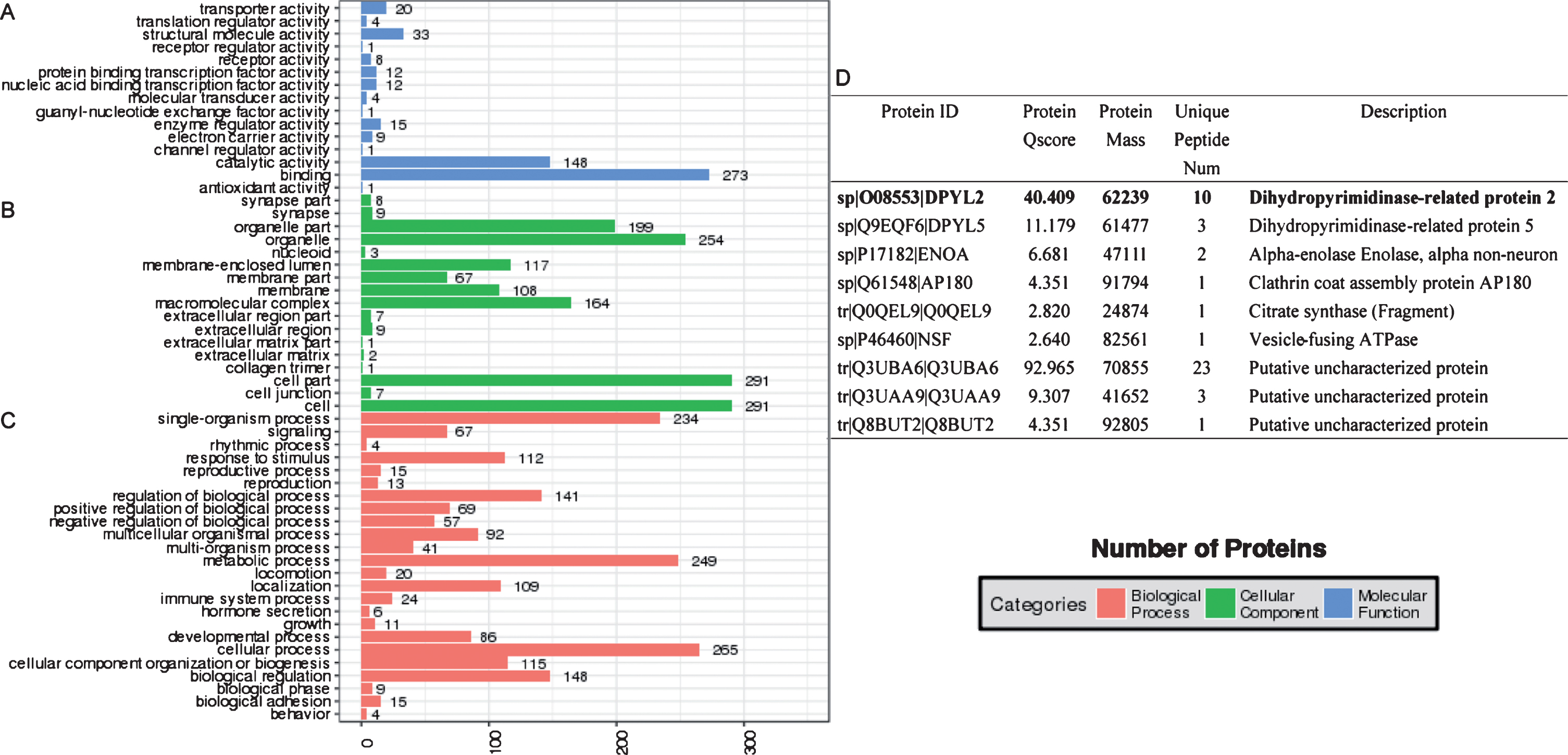
Analysis of gene ontology. Representative GO analysis for the identified proteins interacting with GSK3β in group H3000. GO analysis consisted of molecular function (A), cellular component (B), and biological process (C). Synapse-associated proteins in the cellular component analysis of cells treated with H3000 (D).
The major canonical pathways associated with GSK3β-interacting proteins were enriched by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses (Fig. 4). For the importance of aFGF in neuronal development, we mainly focused on the axon guidance signaling pathway. It was found that CRMP2 was a key protein involved in axon repulsion by regulating microtubule reorganization, which had a high Protein Qscore value in GO cellular component classification (Fig. 3D). It suggested that CRMP2 played an important role in the neuroprotection of aFGF via the signaling pathway mediated by GSK3β.
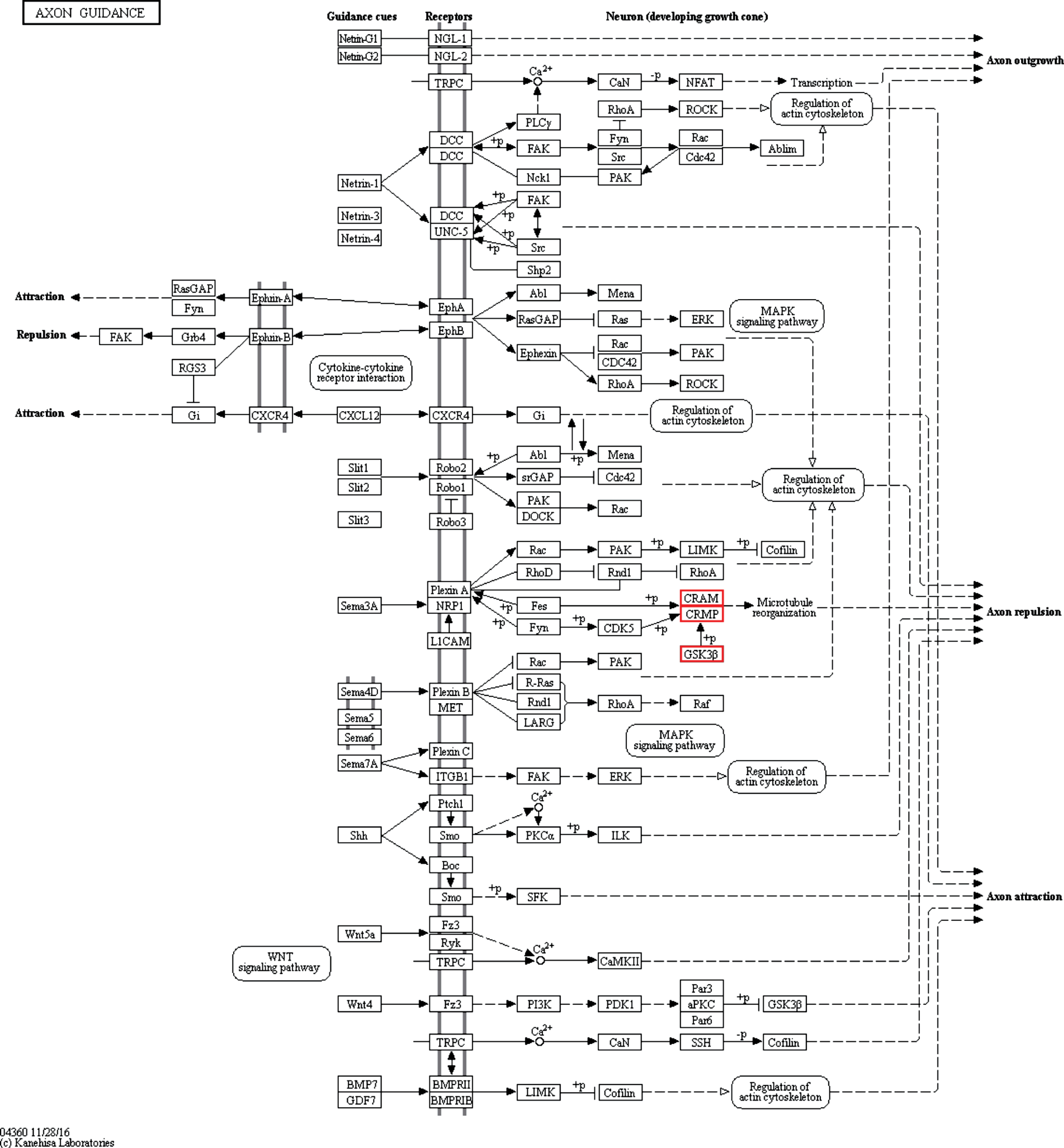
Pathway analysis. Representative KEGG-pathway analysis of axon guidance.
aFGF ameliorated neuron damage induced by Aβ1-42 via GSK3β-CRMP2 signaling pathway
co-IP coupled with immunoblot analysis was performed to validate the interaction between GSK3β and CRMP2. CRMP2 was detectable in the immunoprecipitants pulled down with anti-GSK3β antibody and N2a-APP lysate (input), but not in the immunoprecipitants pulled down with anti-IgG antibody (Fig. 5A). The expressions of CRMP2 and p-CRMP2 (Thr514) were observed in N2a-APP cells treated with different concentrations of aFGF14-154. The phosphorylation level of CRMP2 at Thr514 was significantly reduced by aFGF14-154 in a high concentration (p < 0.01 for H3000 versus Ctrl; Fig. 5B-F). Primary cortical neurons (DIV5) were pre-treated with aFGF14–154 (3000 ng/mL) for 2 h at 37°C, then 8 μM Aβ1-42 was added into the culture medium with continuous aFGF14–154 treatment for 24 h. aFGF14-154 reduced the level of p-CRMP2 (Thr514) and promoted the expression of p-GSK3β (Sre9) without changes in the expression of CRMP2 and GSK3β (p < 0.001 for H+Aβ versus Aβ, p < 0.05 for H+Aβ versus Aβ respectively; Fig. 5G-K).
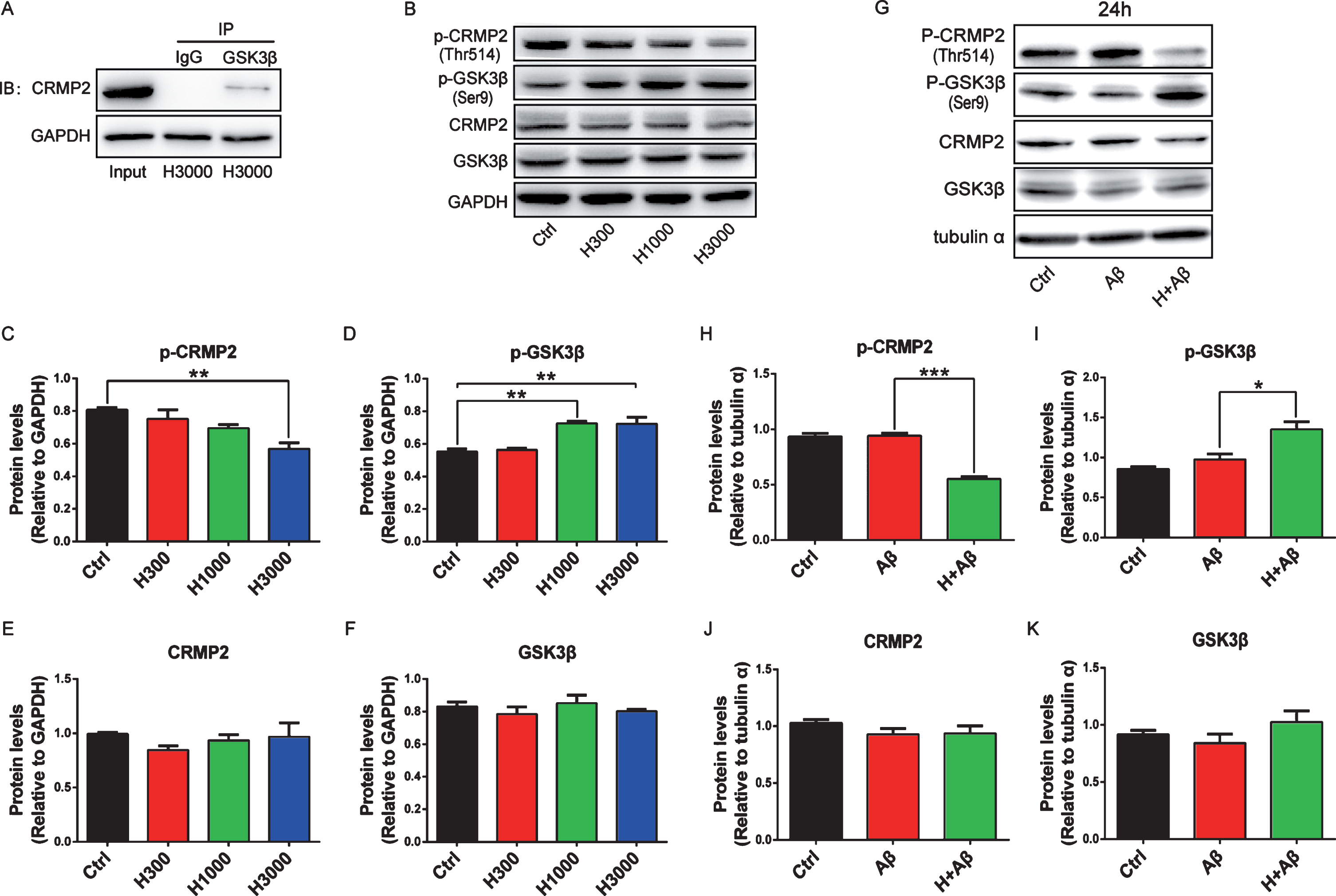
The interaction between GSK3β and CRMP2 was validated. A) CRMP2 was detectable by immunoblotting coupled with co-IP. Representative immunoblots (B) and quantification expression of GSK3β, CRMP2, and their phosphorylation levels in N2a-APP cells treated with aFGF for 24 h (C-F). G-K) Primary cortical neurons (DIV5) were pre-treated with aFGF14–154 (3000 ng/mL-H) for 2 h and 8 μM Aβ1-42 was added into the culture medium with continuous aFGF14–154 incubation for 24 h. Representative immunoblots (G) and quantification expression of CRMP2, GSK3β, and their phosphorylation levels in rat primary cortical neurons damaged by Aβ (H-K). Values were presented as means±SEM (*p < 0.05, **p < 0.01, ***p < 0.001, for significant difference, one-way ANOVA with Bonferroni post hoc test, n = 3).
Neuroprotection of aFGF14-154 is abolished by the knockdown of CRMP2
The small interfering RNA (siRNA) targeting on CRMP2 was used to knock the expression of CRMP2 down in rat primary cortical neurons at DIV5. The knockdown of CRMP2 protein was confirmed by western blotting after the treatments of siRNA for 48 h (p < 0.001 for KD versus NC, Fig. 6A, B). After CRMP2 knockdown, the neuron dendrites were broken and neurite network disappeared with cytoplasmic shrinkage as shown by scanning electron microscopy (Fig. 6C). The average length of neurites decreased significantly (p < 0.01 for KD versus NC, Fig. 6D, E). Normal neurons (H+Aβ) or CRMP2-knockdown neurons (H+Aβ+KD) were treated with aFGF14-154 and Aβ as described above. After the treatment of Aβ, the density of neurites decreased, the axons and dendrites were broken, and the neurite network disappeared, while aFGF14–154 reversed the injury to improve the neurite growth notably. aFGF14–154 markedly increased the average length of neurites in neurons damaged by Aβ, while the amelioration of aFGF14-154 was blocked by the knockdown of CRMP2 (p < 0.05 for H+Aβ+KD versus H+Aβ; Fig. 7A-B). The knockdown of CRMP2 blocked the rescue of aFGF14–154 with broken neurites and shrunken cell bodies in neurons with Aβ injury (Fig. 7C). The effect of aFGF14-154 on enhancing cell viability was abolished after the knockdown of CRMP2 by siRNA in cortical neurons with Aβ1-42 injury (p < 0.05 for H+Aβ+KD versus H+Aβ; Fig. 8A). The synaptic markers synaptophysin (SYN) and postsynaptic density protein 95 (PSD95) were assessed by western blotting to estimate the injury in neurites. The levels of SYN and PSD95 were promoted by Tat-haFGF14-154 in neurons treated by Aβ (p < 0.05). The knockdown of CRMP2 significantly blocked the elevation of SYN induced by aFGF14–154 (p < 0.001 for H+Aβ+KD versus H+Aβ; Fig. 8B-E), but not PSD95.
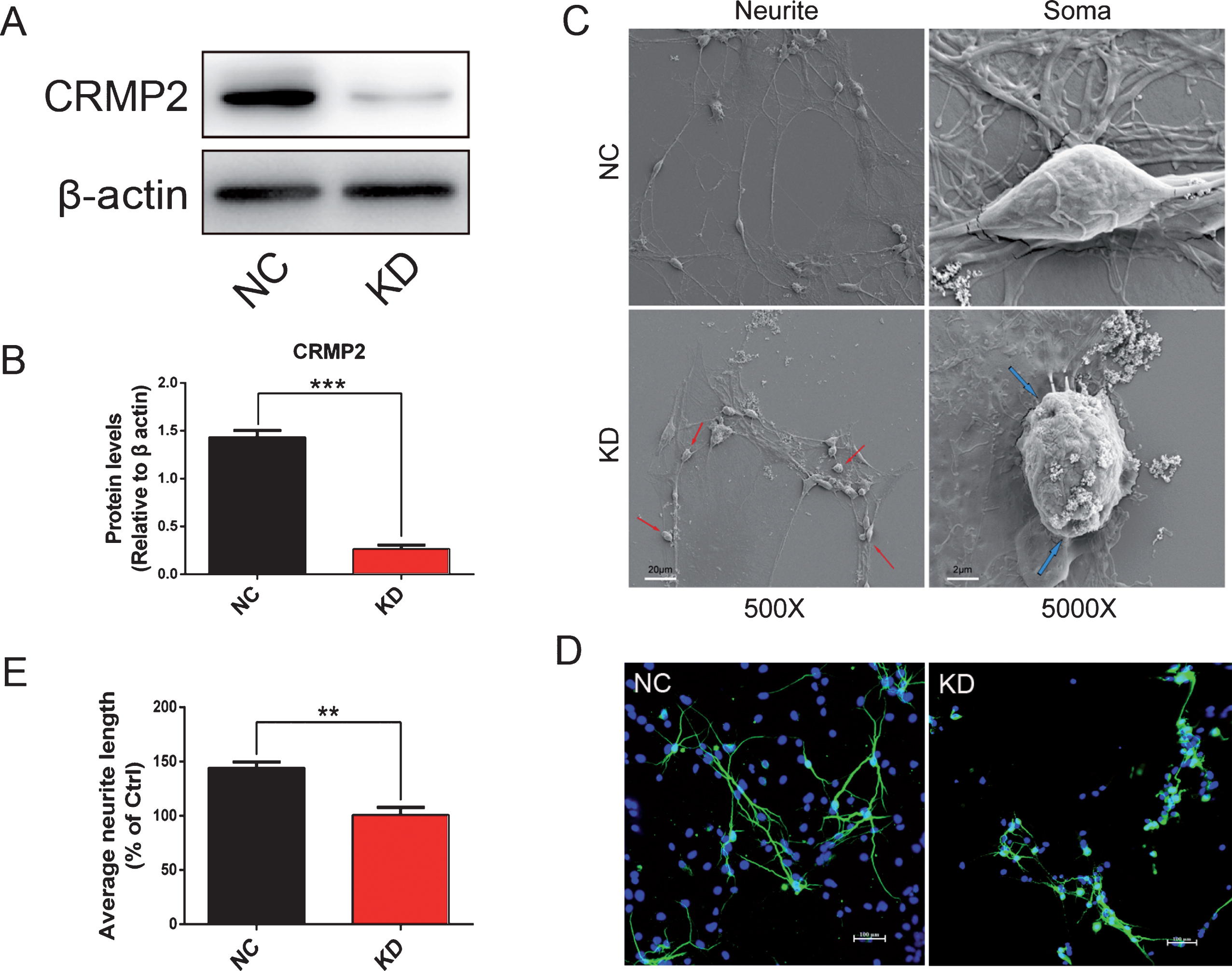
CRMP2 was knocked down by siRNA in rat cortical neurons. The neurons were transfected with targeted-siRNA of CRMP2 (KD) or scrambled-siRNA (NC) for 24 h. The expression of CRMP2 was significantly reduced by the knockdown of siRNA (A, B). The soma and neurites of neurons were observed by scanning electron microscopy (C). The damaged neurites are shown by red arrows and the shrunken cell bodies are shown by blue arrows. Representative immunofluorescence images of neurons with MAP2-positive staining (scale bar: 20 μm) and the quantification of neurites length (D, E). The density of neurites decreased, axons and dendrites were broken, and neurite network disappeared with decreased neurite length in cortical neurons after CRMP2 knockdown. Values were presented as means±SEM (**p < 0.01, ***p < 0.001 for significant difference, one-way ANOVA with Bonferroni post hoc test, n = 3).
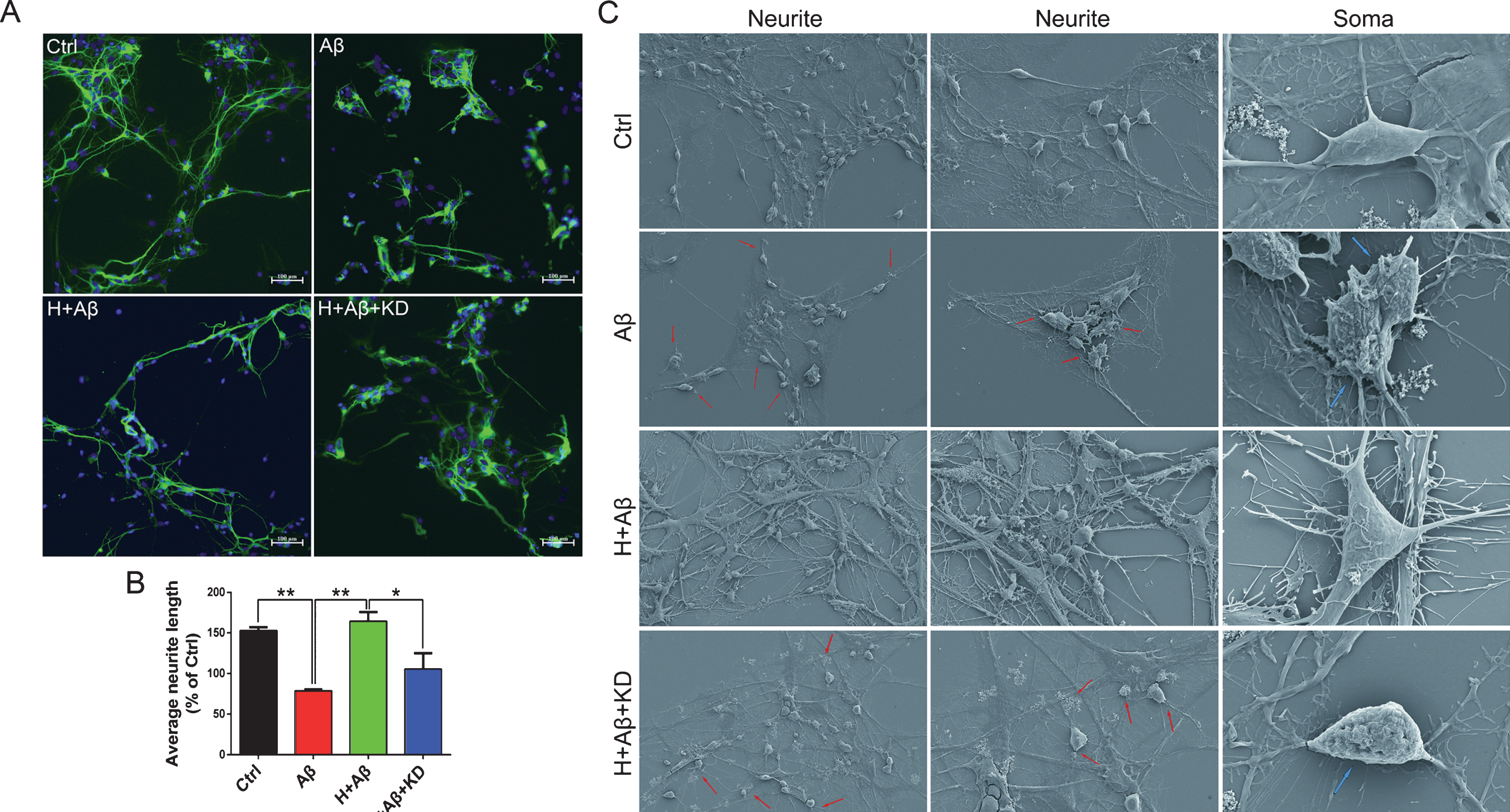
The knockdown of CRMP2 blocked the rescue of aFGF14–154 in rat primary cortical neurons damaged by Aβ. A) Representative immunofluorescence images of neurons with MAP2-positive staining (scale bar: 20 μm). B) The quantification of neurites length. The soma and neurites of neurons were observed by scanning electron microscopy (C). The damaged neurites are shown by red arrows and the shrunken cell bodies are shown by blue arrows. The knockdown of CRMP2 blocked the rescue of aFGF14–154 with broken neurites and shrunken cell bodies in neurons with Aβ injury. aFGF14–154 markedly increased the average length of neurites in neurons damaged by Aβ, while the amelioration of aFGF14-154 was blocked by the knockdown of CRMP2. Values were presented as means±SEM (*p<0.05, **p < 0.01 for significant difference, one-way ANOVA with Bonferroni post hoc test, n = 3).
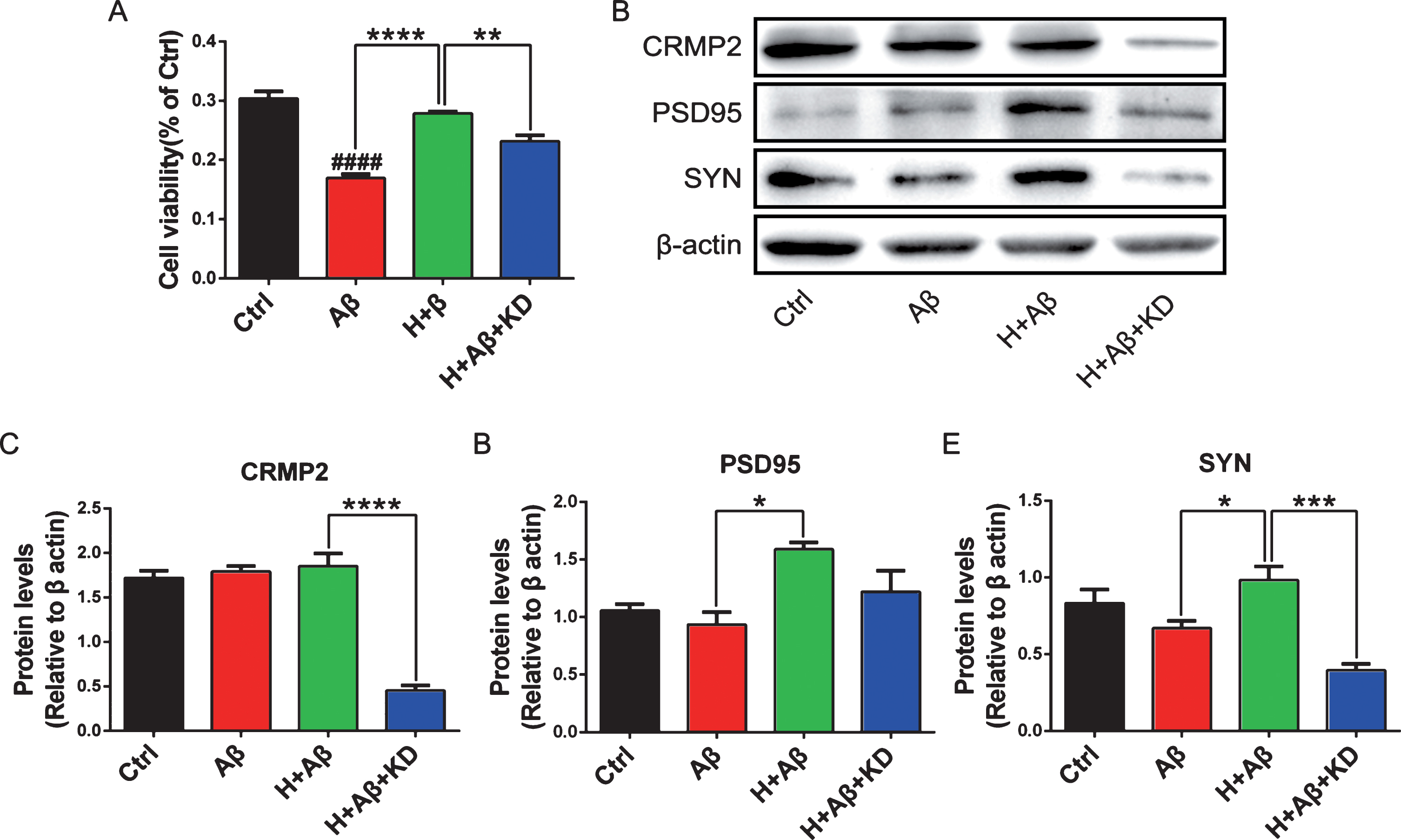
The knockdown of CRMP2 abolished the elevation of SYN induced by aFGF14–154. Rat primary cortical neurons were treated with or without Aβ1-42/aFGF14–154/siRNA as described above. A) Cell viability detected by MTT assay. Representative immunoblots (B) and quantification expression of CRMP2 (C), PSD95 (D), and SYN (E). Values were presented as means±SEM (*p < 0.05, ***p < 0.001, ****p < 0.001 for significant difference, one-way ANOVA with Bonferroni post hoc test, n = 3).
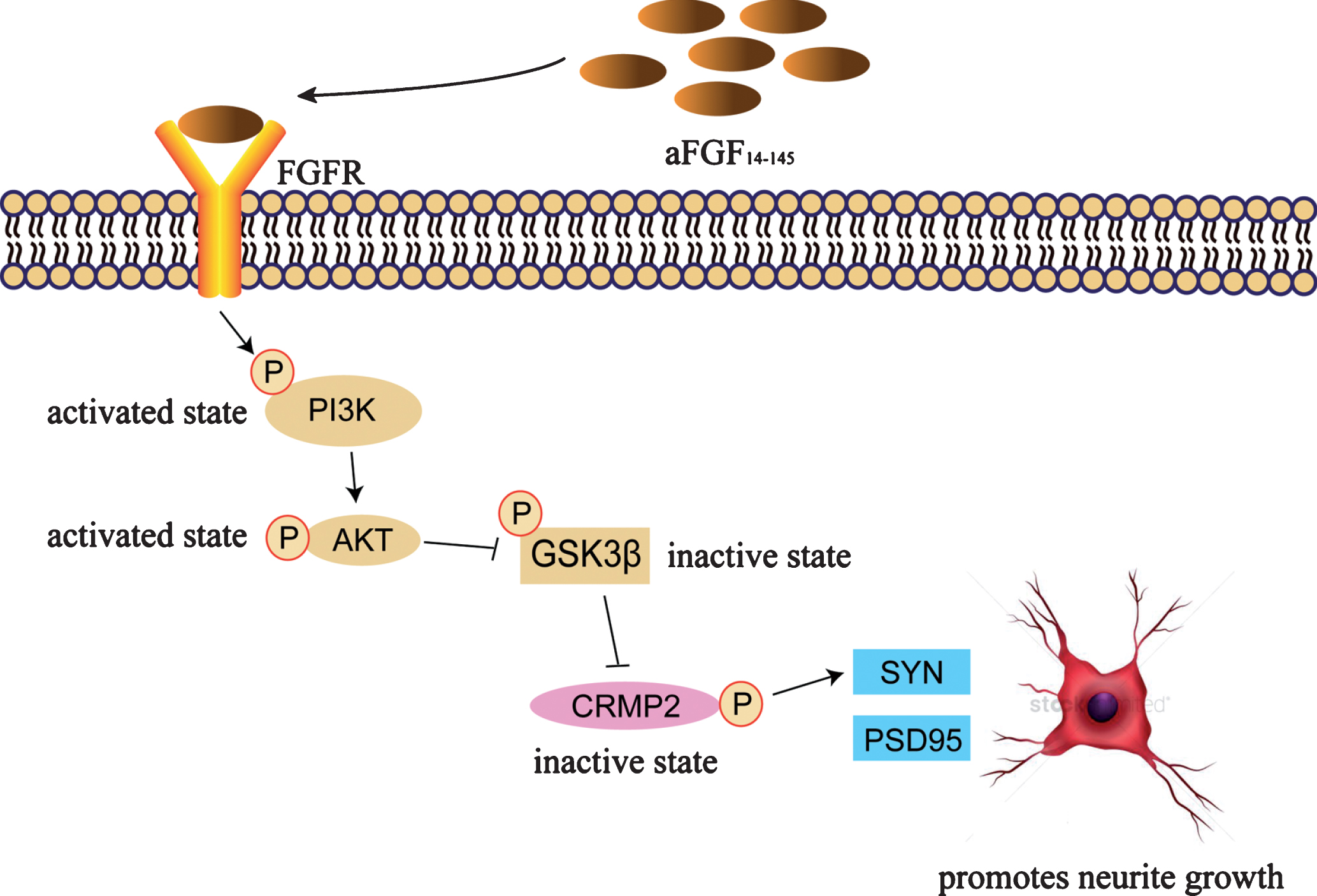
aFGF promotes neurite growth via GSK3β-CRMP2 signaling pathway. aFGF14-154 binds to its receptor, activates the PI3K pathway, followed by an inhibition in the activity of GSK3β with an increase of the phosphorylation at Ser9. The upregulated phosphorylation of GSK3β inhibits the phosphorylation of CRMP2 (Thr514) to enhance its activity, which leads to the neurite growth.
DISCUSSION
In this study, aFGF14-154 increased the phosphorylation of GSK3β (Ser9) both in N2a-APP cells and rat primary cortical neurons damaged by Aβ1-42, thereby to inhibit the activity of GSK3β. aFGF14-154 significantly increased the average neurite length of neurons damaged by Aβ1-42, which was reversed by the inhibition of GSK3β. This suggests that GSK3β is related to the amelioration of aFGF in neurons damaged by Aβ. When spinocerebellar ataxia type 1 model mice were fed a diet containing GSK3β inhibitor (0.2% lithium carbonate), motor coordination, learning, and memory skills improved significantly, while GSK3β inhibitor alleviated the reduction in dendritic branching of mutant hippocampal pyramidal neurons [24]. GSK3β inhibitors have potential as therapeutic target for cognitive dysfunction, and there are many preclinical studies providing evidences in restoring cognitive function [25].
It is still unknown which downstream substrates of GSK3β are involved in promoting neurite growth of aFGF14-154. Many GSK3β substrates are microtubule-associated proteins bound to neurite growth, including APC, CRMP2, MAP1B, microtubule associated protein 2 (MAP2), tau, neurofilament, and kinesin light chain [26]. The proteins interacting with GSK3β were detected by co-IP and LTQ-Orbitrap proteomics technology to screen the downstream substrates. CRMP2 interacting with GSK3β was found to play an important role in the neuroprotection of aFGF.
CRMP2, also known as DRP2, is highly expressed in the central nervous system. It is first discovered that the biological function of CRMP2 is to guide axon formation and growth cone collapse, while overexpression of CRMP2 can accelerate neurons to regenerate [27]. The CRMP2 knockdown decreases growth of CRMP2-dependent neurite in cortical neurons, and CRMP2 promotes the oligomerization of tubulin dimers by binding to tubulinα/β to improve microtubules aggregation and stabilization in vitro [27]. CRMPs play an important role in the pathogenesis of nervous system diseases, including AD, Parkinson’s disease, schizophrenia, and epilepsy. The anti-epileptic drug-lacamide, which targets CRMP2, relieves neurodegenerative symptoms [28]. Brain-specific CRMP2 deletion leads to neuronal development deficits and behavioral impairments in mice; GluN1 and GluN2B which are receptor subunits of SYN and N-methyl-D-aspartate (NMDA) reduce significantly in the hippocampus, and the complexity, density, and volume of pyramidal neuron dendrites also decrease [29]. In consistence with these reports, we found that the neurite length of cortical neurons was decreased and the dendrites were broken with cytoplasmic shrinkage after CRMP2 knockdown by siRNA.
GSK3β phosphorylates CRMP2 at Thr509/514 after cyclin-dependent kinase 5 (Cdk5) pre-phosphorylated at Ser522, which contributes to the microtubule destabilization [30, 31]. Genetic inhibition of GSK3β reduces the phosphorylation of CRMP2 and improves optic nerve regeneration; CRMP2 inhibition abrogates neurite growth promotion and myelin disinhibition mediated by GSK3β knockout [32]. aFGF14-154 inhibited the activity of GSK3β by upregulating the phosphorylation of GSK3β (Ser9), so it decreased the phosphorylation of CRMP2 (Thr514) to enhance the activity of CRMP2. Increased CRMP2 phosphorylation is apparent in the brains of AD patients and mouse models that express mutated APP and PS1, but not in cultured neurons damaged by oligomeric Aβ [33]. We have obtained similar experimental results in our study by showing that Aβ does not induce the phosphorylation of CRMP2. In cortical neurons damaged by Aβ, aFGF14-154 upregulated the expression of SYN and PSD95, and then promoted neurite growth; the knockdown of CRMP2 by siRNA blocked the growth of neurites and the upregulation of SYN induced by aFGF14-154. It suggests that aFGF14-154 promotes neurite growth by inhibiting CRMP2 phosphorylation.
In conclusion, CRMP2 has been identified as a protein interacting with GSK3β by co-IP coupled with LTQ-Orbitrap proteomics technology, which is involved in the axon repulsion by regulating microtubule reorganization. aFGF14-154 increased the phosphorylation of GSK3β (Ser9) to inhibit its activity, then followed by a low phosphorylation level of CRMP2 (Thr514), which leads to the neurite growth. These results highlighted the important role of CRMP2 and its phosphorylation through GSK3β in the effect that aFGF14-154 promoted the growth of neurite damaged by Aβ1-42.