
Abstract
Background:
Mild cognitive impairment (MCI) has been considered as a pre-dementia stage, although the factors leading to Alzheimer’s disease (AD) conversion remain controversial.
Objective:
Evaluate whether TOMM40 poly-T (TOMM40′ 523) polymorphism is associated with the risk and conversion time from MCI to AD and secondly with AD cerebrospinal fluid (CSF) biomarkers, disentangling the APOE genotype.
Methods:
147 AD patients, 102 MCI patients, and 105 cognitively normal controls were genotyped for poly-T polymorphism. MCI patients were subdivided into two groups, the group of patients that converted to AD (MCI-AD) and the group of those that remained stable (MCI-S).
Results:
TOMM40′ 523 L allele was significantly more frequent in the MCI-AD group and having at least one L allele significantly increased the risk of conversion from MCI to AD (OR = 8.346, p < 0.001, 95% CI: 2.830 to 24.617). However, when adjusted for the presence of APOE ɛ4 allele, both the L allele and ɛ4 allele lost significance in the model (p > 0.05). We then analyzed the APOE ɛ4-TOMM40′ 523 L haplotype and observed that patients carrying this haplotype had significantly higher risk (OR = 5.83; 95% CI = 2.30–14.83) and mean lower times of conversion to AD (p = 0.003). This haplotype was also significantly associated with a biomarker profile compatible with AD (p = 0.007).
Conclusion:
This study shows that the APOE ɛ4-TOMM40′ 523 L haplotype is associated with a higher risk and shorter times of conversion from MCI to AD, possibly driven by CSF biomarkers and mitochondrial dysfunction.
Keywords
INTRODUCTION
The term “mild cognitive impairment” (MCI) describes an intermediate state of cognitive function between the changes seen in aging and those fulfilling the criteria for dementia, namely Alzheimer’s disease (AD) [1]. The rate of conversion from MCI to AD is estimated to reach 10–15% per year in contrast to a rate of 1–2% per year among healthy elderly individuals [2]. When considering MCI as a prodromal state of AD (MCI due to AD) [3], it is of major importance to study the risk factors that predict which MCI patients will convert to AD, namely risk genes, as this group of patients represents a target for future disease modifying therapies [4].
The ɛ4 allele of Apolipoprotein E gene (APOE), the most highly replicated genetic risk factor for late-onset AD (LOAD) [5, 6], has been shown to increase the susceptibility for MCI to AD progression, as demonstrated in previous meta-analysis [7, 8]. However, the presence of an allele ɛ4 is neither necessary nor sufficient for MCI to AD progression, showing low sensitivity. Moreover, its use for MCI to AD prediction remains controversial. For this reason, recent models of MCI to AD progression incorporate other predictors besides allele ɛ4, such as: cerebrospinal fluid (CSF) biomarkers, neuroimaging biomarkers (e.g., magnetic resonance imaging (MRI) and fluorodeoxyglucose positron emission tomography (FDG-PET)) and neuropsychological tests, in order to increase the accuracy when evaluating the conversion from MCI to AD [9–11]. Apart from APOE, there is little information about genes that increase the susceptibility for MCI to AD progression. Interestingly, the most relevant discoveries in fine-mapping and genome-wide association studies for AD are within a linkage disequilibrium (LD) region in Chr:19q13.32 that incorporates APOE, TOMM40, and APOC1 genes [12–14]. By deep sequencing of this specific region, Roses’ group discovered a poly-T polymorphism, rs10524523, in the translocase of outer mitochondrial membrane 40 homolog (TOMM40) gene (hereafter, TOMM40′ 523), reported to be associated with risk and age of onset of AD [15]. Meanwhile, based on the distribution behavior of the number of thymine (T) residues, three categories (alleles) of repeat length were established: short (S, ≤19), long (L, 20–29), and very long (VL, ≥30) [16]. In the Caucasian population, it has been confirmed that the APOE ɛ4 allele is almost exclusively linked to a L poly-T repeat allele [15, 16]. This strong LD (r2 = 0.941) [17] between TOMM40′ 523 L and APOE ɛ4 alleles complicates analytical approaches to disentangle independent effects between these two variants as fleshed out in previous studies [18–20]. Nevertheless, these effects could be due to a combination of both APOE and TOMM40 [17]. As the APOE ɛ3 allele is almost always linked to either a S or a VL poly-T allele, in the Caucasian population, several studies attempted to find instead differences between S and VL alleles in ɛ3/ɛ4 or ɛ3/ɛ3 cohorts in order to establish an effect of TOMM40′ 523 independent of APOE. However, differences between these two alleles (S and VL) are still controversial (reviewed in Chiba-Falek et al.) [21].
Unlike the APOE ɛ4 allele, the role of TOMM40′ 523 has been poorly addressed in MCI cohorts. One study presented a stratification by TOMM40′ 523 and APOE genotype of the age of onset of cognitive impairment for cognitively normal subjects followed in a prospective cohort, in order to create a risk algorithm for clinical trial enrichment [22], which was further evaluated in other studies [23, 24]. A subsequent study evaluated, the utility of TOMM40 poly-T variable-length polymorphism alleles among other 247 variables, for modelling progression from MCI to AD [25]. Lastly, an additional study reported the effect of TOMM40′ 523 on spatial navigation in amnestic MCI individuals [26]. However, as far as we know, none of these studies fully addressed the connection between TOMM40′ 523 polymorphism and the risk and time of conversion from MCI to AD.
The main aim of this study was to investigate the relation between TOMM40′ 523 polymorphism with the risk and conversion time from MCI to AD, and to disentangle the effect of APOE genotype in that relation. For this purpose, the poly-T profile was first characterized in three different groups: cognitively normal controls, AD and MCI patients subdivided into patients that converted to AD (MCI-AD), and patients that remained stable (MCI-S) within a minimum period of two years. Secondly, the association between TOMM40′ 523/APOE genotype and the AD CSF biomarkers, particularly amyloid-β 42 (Aβ42), total, and phosphorylated tau (t-tau and p-tau) was also explored.
MATERIALS AND METHODS
Study subjects
This study included 354 Caucasian subjects: 147 AD patients, 102 MCI patients, and 105 cognitively normal controls. CSF and DNA from AD and MCI individuals were available, DNA samples were also available from controls.
AD and MCI patients were recruited and diagnosed at the Dementia Clinic, Neurology Department of Coimbra University Hospital (CHUC), Coimbra, Portugal. The baseline study, as well as the follow-up protocol, have already been published [27]. In summary, patients were enrolled in a systematic way and were subject to biannual clinical observation and annual neuropsychological and functional evaluations. All patients undertook a thorough biochemical, neurological, and imaging (computed tomography (CT) or MRI) evaluation. PET and genetic studies were more restricted, although considered in younger patients. Essentially, a neurologist completed a medical history with the patient and caregiver and conducted a general physical, neurological, and psychiatric examination in addition to a comprehensive battery-protocol diagnostic, including cognitive instruments. Cognitive instruments included: the Portuguese versions, validated for the Portuguese population, of the Mini-Mental State Examination (MMSE) [28, 29], the Montreal Cognitive Assessment (MoCA) [30, 31], and the Alzheimer’s Disease Assessment Scale-Cognitive (ADAS-Cog) [32–34]. A comprehensive neuropsychological battery with normative data for the Portuguese population (Lisbon Battery for Dementia Assessment) [35] exploring memory (Wechsler Memory Scale subtests) and other cognitive domains (including language, praxis, executive functions, and visuoconstructive tests) were also used. The evaluation was completed applying standard staging scales which provided objective information about subject performance in various domains, including the Clinical Dementia Rating scale (CDR) [36] for global staging, the Disability Assessment for Dementia (DAD) [37, 38] to evaluate the functional status, and, the Neuropsychiatric Inventory (NPI) [39, 40] to characterize the psychopathological profile, including the presence of depression. All the available information (baseline cognitive test, staging scales, clinical laboratory, and imaging studies) was used to reach a consensus research diagnosis. A similar approach was used to follow-up annual evaluations.
MCI patients were amnestic type (aMCI) and the diagnosis was completed in agreement with the criteria defined by Petersen et al. [41] and, more recently, with the framework for MCI due to AD proposed by NIA-AA criteria [3]. Petersen et al.’s criteria were implemented as follows: 1) A subjective complaint of memory decline (reported by the subject or informant); 2) An objective memory impairment (considered when scores on standard Wechsler memory tests were >1.5 SDs below age/education adjusted norms) with or without deficits in other cognitive domains; 3) Normal general cognition suggested by normal scores in the MMSE and MoCA using the Portuguese cut-off scores [29, 31]; 4) Fairly normal daily life activities, evaluated with a functional scale –(DAD) 5) Absence of dementia, indicated by a CDR rating of 0.5 [36]. All patients were in stable condition without acute comorbidities. As exclusion criteria for enrolment, we considered a significant underlying medical or neurological illness revealed by laboratory tests or imaging; a relevant psychiatric disease, including major depression, suggested in the medical interview and confirmed by the NPI; and CT or MRI demonstration of significant vascular burden [42].
MCI cases were followed, at least for 2 years, under this comprehensive protocol and they were further dichotomized in those that were cognitively stable (MCI-S) and those that developed dementia due to AD (MCI-AD). Conversion to AD required fulfilling the criteria of the clinical diagnostic for probable AD (see below) and was confirmed by the coordinator of the clinical study (IS). As these criteria are not fully operational and the conversion status decision has some uncertainty and subjectivity, patients in this study were classified as having undergone conversion based on: 1) Objective evidence by cognitive testing of deterioration to dementia using the MMSE, the MoCA, and the ADAS-COG scores and qualitative evaluation (i.e., impairment of memory with the addition of other domains); 2) Changes in global CDR rating from 0.5 to 1 or more, confirming the cognitive profile of dementia and autonomy loss. Dementia was diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders –fourth edition text review (DSM-IV-TR) criteria [43], and AD, according to the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders (NINCDS-ADRDA) [44] and, more recently, the 2011 NIA-AA criteria [45]
Cognitively normal controls (≥65 years) were recruited from a group of volunteers from a Portuguese population study on aging [46], who were subjected to cognitive evaluation and showed no signs of cognitive impairment. These procedures have been approved by the Ethics Board of CHUC and Faculty of Medicine of Coimbra. Informed consent was required from all subjects or responsible caregivers, whatever appropriate.
APOE and TOMM40 genotyping
DNA was isolated from whole EDTA-blood using a commercial kit (Roche Diagnostics GmbH, Manheim, Germany). APOE genotype was determined by polymerase chain reaction-restriction fragment length polymorphisms (PCR-RFLP) assay, as previously described [47] TOMM40′ 523 polymorphism was genotyped as described formerly [48], with some modifications. Briefly, each genomic DNA sample was amplified by PCR using fluorescently labelled forward 5′D4-TGCTGACCTCAAGCTGTCCTC-3′ and reverse 5′-GAGGCTGAGAAGGGAGGATT-3′ primers. PCR amplification was performed in a 12μL volume containing 20 ng of genomic DNA, 2.5μM of each primer, and 6μl of Supreme NZYTaq 2× Green MasterMix (Nzytech, Lisbon, Portugal). Amplification was performed, with the following conditions: initial denaturation for 5 min at 96°C; 29 cycles of denaturing for 45s at 96°C, annealing for 45 s at 69°C and elongation for 45s at 72°; concluded with a single 10 min final elongation step at 72°C. PCR products with an expected length of 150 + n(T) pb were confirmed in a 2% agarose gel electrophoresis. Afterwards, 1μl of each PCR product was mixed up with 20μl of formamide (Beckam Coulter, USA) and 0.5μl of DNA Size Standard 400 (Beckam Coulter, USA) and loaded on a capillary automated sequencer CEQ 8000 (Beckman Coulter, USA).
Genotype calling
Since we were analyzing a homopolymer, “slippage” during each DNA amplification was observed leading to a final product of amplicons with variable lengths around the real poli-T length. However, as previously described [48, 49], the PCR amplicons have a normal distribution with maximum intensity/area of signal near the true count of T (N[T]), but distributed continuously with error around the true value (Fig. 1). Genotypes were determined using the tool Fragments of the Genome Lab™ software version 10.2 (Beckman Coulter, USA) under the presumption that the highest area peak corresponded to the true count of “Ts”. This analysis was always performed by two independent observers who categorized the poly –T fragments according to the poly-T lengths into: short (S, ≤19), long (L, 20–29), or very long (VL, ≥30) repeats as previously described [16]. In order to validate and compare the length of poly-T repeats, we sequenced 6 samples by Sanger sequencing and compared the poly-T size with the one determined from the fragment analysis. The results obtained by both techniques were similar, with an estimated error of +/–1bp.
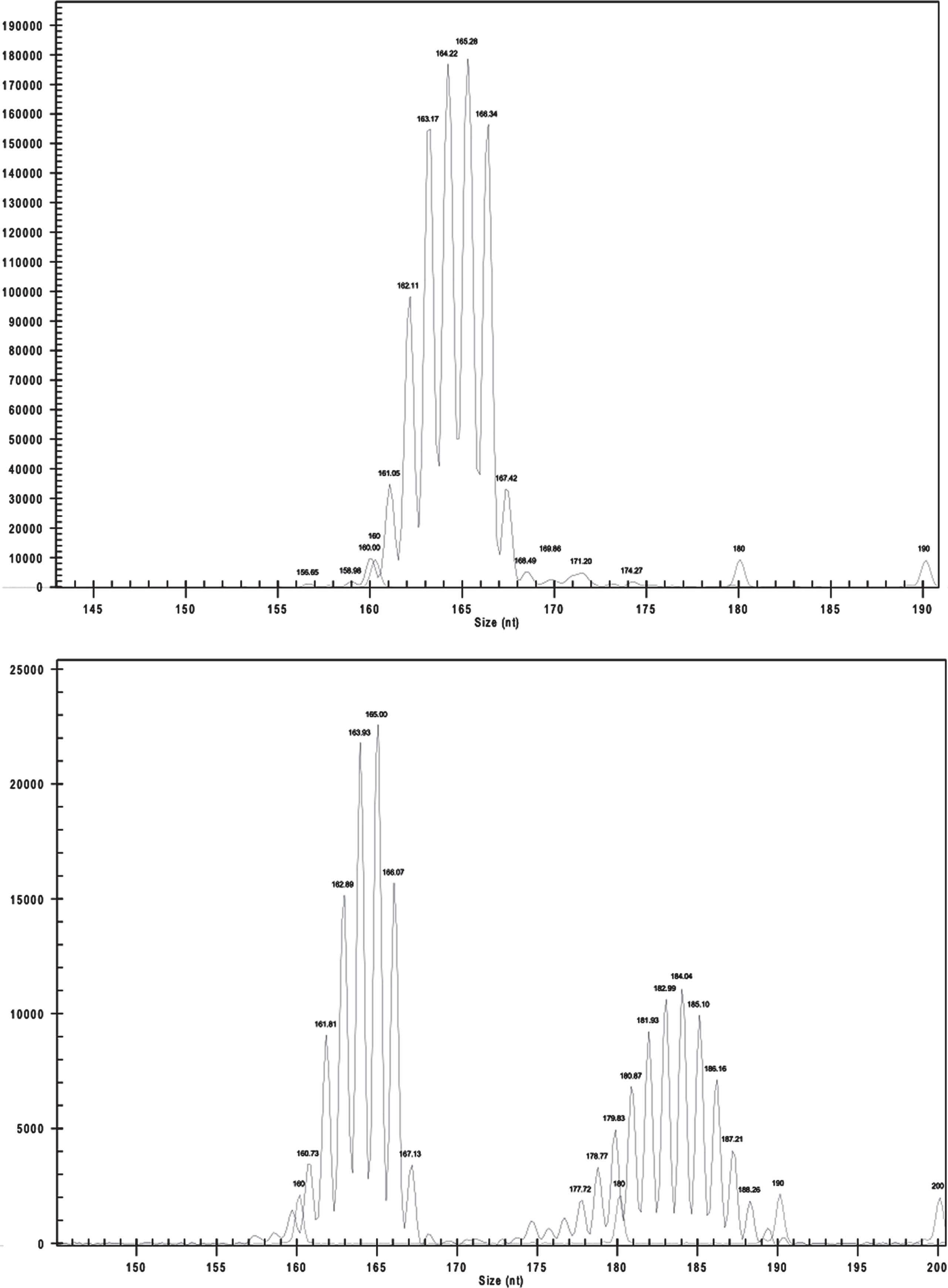
Count of polymerase chain reaction (PCR) fragments (intensity) versus size of fragment of a patient homozygous for TOMM40′ 523 allele (S/S) (up) and patient heterozygous for TOMM40′ 523 allele (S/VL) (down). The PCR amplicons have a normal distribution with maximum intensity of signal near the true count of T (N[T]).
Cerebrospinal fluid biomarkers analysis
CSF samples were collected from patients as part of their clinical diagnosis investigation routine. Pre-analytical and analytical procedures were done in accordance with previously published recommendations [50]. Briefly, CSF samples were collected into sterile polypropylene tubes, immediately centrifuged at 1800 g for 10 min at 4°C, aliquoted into polypropylene tubes and stored at –80°C until analyzed. CSF Aβ42, t-Tau, and p-Tau-181 levels were measured separately, via commercially available sandwich ELISA kits (Innotest; Innogenetics/Fujirebio, Ghent, Belgium), as aforesaid [51]. All samples were tested in duplicate and assays were performed sequentially in a clinical routine setting. External quality control of the assays was completed under the scope of the Alzheimer’s Association Quality Control Program for CSF Biomarkers [52]. The CSF biomarkers profile was classified using the Innotest Amyloid-Tau Index (IATI), with the following formula: Aβ/[240 + (1.18 × Tau)] , and a biomarker profile compatible with AD was defined with a score below 1 [53].
Statistical analysis
Categorical data was presented in observed counts and percentages, whereas the mean, standard deviation, and range were used for quantitative data. Group differences concerning qualitative data were analyzed through the Chi-square test, adjusted for pairwise comparisons using a Bonferroni correction whenever justified, while non-parametric Mann-Whitney and Kruskal Wallis were applied for comparisons between groups in quantitative data. Also, the association between the haplotype and risk for conversion was also assessed by a Chi-Square test. Z test adjusted with the Bonferroni correction was used to compare the relative frequencies of poly-T lengths (S, L, or VL) among different diagnosis groups. Logistic regression was applied to assess the risk of conversion in MCI patients. Kaplan-Meyer survival analysis was used to search for predictors in time to conversion in MCI patients. Both analyses were adjusted for confounding factors such as gender, age, and APOE genotype, specifically the ɛ4 allele. All tests were analyzed at a significance level of 5 %. Statistical analysis was conducted in SPSS, version 25.
RESULTS
Sample characteristics
Demographic and clinical characteristics of the 354 participants involved in this study, as well as comparisons by diagnostic group, are summarized in Table 1. No differences were found, between groups, in gender distribution (p = 0.097). Regarding age, it was observed that controls and MCI-AD patients were significantly older than the remaining groups (p < 0.001). Similarly, we observed that MCI-AD patients were significantly older than AD and MCI-S groups regarding age of onset (p < 0.001). AD and MCI-AD patients had a significant higher percentage of APOE ɛ4 allele carriers (45.6% and 59.7%, respectively) than MCI-S and controls (20% and 18.1%, respectively) (p < 0.001). Concerning CSF-AD biomarkers, AD and MCI-AD had significantly lower Aβ42 levels and significantly higher t-tau and p-tau levels than MCI-S (p < 0.001).
Demographics and clinical characteristics of the studied cohort
Data is presented as mean±SD, except when indicated otherwise.
TOMM40 poly-T distribution
In all clinical groups, we observed that the distribution frequency of TOMM40 poly-T lengths had several peaks in the fragment analysis, consistent with other published studies with European cohorts [20, 54], confirming our TOMM40′ 523 genotyping methodology. Particularly, 4 clusters with peaks around 15, 22, 28, and 33/34 “Ts” with diminished count frequencies on either side of the peak, could be distinguished (Fig. 2). When comparing MCI-S and MCI-AD distribution of poly-T lengths, it was observed that despite an overall similar pattern, differences between poly-T relative frequencies could be seen in these groups (Fig. 2A). The same was observed when comparing controls and AD poly-T lengths distribution (Fig. 2B). In order to evaluate if these differences were statistically significant, we compared the relative frequencies of poly-T lengths classified as S, L, or VL among different diagnosis groups using a Z test adjusted with the Bonferroni correction (Table 2). By comparing MCI-AD with MCI-S group we observed that MCI-AD group had significantly higher frequencies of L alleles (p < 0.001) than MCI-S group. Regarding S and VL alleles, MCI-AD group had lower relative frequencies of S and VL alleles than MCI-S group. However, these differences did not reach statistical significance (p > 0.05) after Bonferroni correction (Table 2). Similar results were observed when comparing AD versus controls. Therefore, this data suggests that, in general MCI-AD is similar to AD and MCI-S to controls regarding TOMM40′ 523 allele distribution (Table 2).
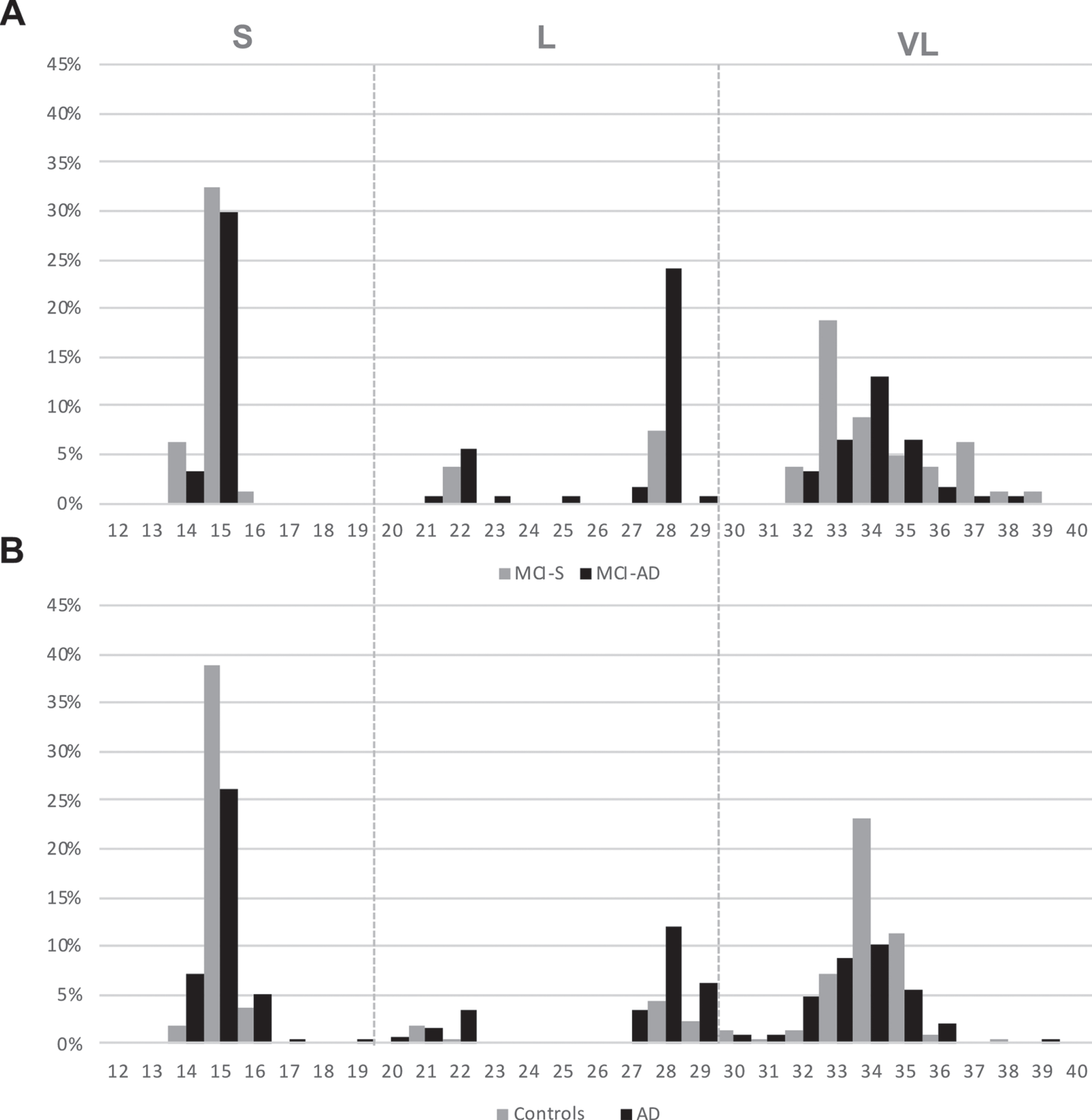
Distribution of TOMM40′ 523 poly-T lengths (relative frequencies) in MCI-S versus MCI-AD group (A) and controls versus AD group (B). TOMM40′ 523 alleles are classified into: short (S, ≤19), long (L, 20–29), or very long (VL, ≥30).
Distribution of TOMM40 poly-T alleles in the AD, MCI-AD, MCI-S, and control groups
Data is presented as relative frequencies of TOMM40 poly-T alleles (S, L, and VL) in each clinical group. Statistical analysis was performed using Z test adjusted for pairwise comparisons using a Bonferroni adjustment.; # # #p < 0.001 versus AD; # #p < 0.01 versus AD; #p < 0.05 versus AD; ‡‡‡p< 0.001 versus MCI-AD.
The different TOMM40′ 523 genotypes, for MCI population and its correlation with APOE genotype are shown in Table 3. Here, we observed that in both MCI-S and MCI-AD population, the TOMM40′ 523 genotype was significantly associated to APOE (p < 0.001). APOE ɛ3 allele was found to be associated to TOMM40′ 523 S and VL alleles, whereas the ɛ4 allele was almost exclusively associated to L TOMM40′ 523, as previously described [15, 16]. Despite the low frequency of APOE ɛ2 allele carriers, we found that similar to the ɛ3 allele, the ɛ2 alleles were also associated with the S and VL alleles. However, some discrepancies were found and are underlined and highlighted in bold in Table 3. The same association was found in the control and AD group (data not shown).
Distribution of TOMM40’ 523 polymorphism according to apolipoprotein E (APOE) genotype for MCI-S and MCI-AD population
Significantly statistic associations (Fisher test) between APOE genotypes and TOMM40′ 523 genotypes are highlighted in dark grey. Discrepancies are underlined and highlighted in bold.
TOMM40′ 523 genotypes and risk of MCI conversion to AD
We performed a logistic regression analysis in order to assess the risk conferred by the different TOMM40′ 523 genotypes for MCI to AD conversion. In a preliminary analysis, we observed that TOMM40 ′523 genotype was significantly associated with the risk of MCI to AD conversion and more specifically that TOMM40′ 523 genotypes S/L and L/L were associated with that risk. Curiously, the L allele, when combined with the VL allele (L/VL) did not reach statistical significance (data not shown). However, when we stratified by TOMM40′ 523 genotypes, the sample size was very small within each stratum, therefore we considered this analysis only as preliminary, due to low statistical power.
We then analyzed the impact of TOMM40′ 523 L allele on MCI to AD risk of conversion and observed that having at least one L allele significantly increased the risk of conversion (OR = 8.346, p < 0.001, 95% CI: 2.830 to 24.617). Age also showed to modestly impact the risk of conversion (OR = 1.140, p < 0.001, 95% CI: 1.064 to 1.221) but gender was not a significant factor (p > 0.05). However, as demonstrated previously, the L allele is almost exclusively linked to APOE ɛ4 allele, which also demonstrated to increase the risk of conversion (OR = 9.033, p < 0.001, 95% CI: 3.055 to 26.709). When we adjusted for ɛ4 allele, as expected, the L allele and ɛ4 allele were not independent significant factors for the risk of conversion (Table 4).
Association of TOMM40′523 L and APOE ɛ4 with risk of MCI conversion to AD
APOE ɛ4-TOMM40′ 523 L haplotype on risk and time of conversion
As it was not possible to disentangle if the effect would be due to ɛ4 allele or due to TOMM40′ 523 L allele, we analyzed instead the APOE ɛ4-TOMM40′ 523 L haplotype (hereafter ɛ4-L haplotype) since L allele in our sample occurs in 95.5% of ɛ4 carriers. We then performed a haplotypic analysis, where we compared the carriers of the ɛ4-L haplotype with those not carrying that haplotype and observed a significantly higher risk of conversion for the haplotype carriers (OR = 5.83; 95% CI = 2.30–14.83), p < 0.001. When evaluating the impact of the ɛ4-L haplotype on the time of conversion of MCI patients to AD, we also observed that carriers of this haplotype had significantly lower mean times of conversion: 4.8 years (95% CI: 3.7 to 5.9) than patients without the ɛ4-L haplotype: 9.1 years (95% CI: 7 to 11.1) (p = 0.003) (Fig. 3).
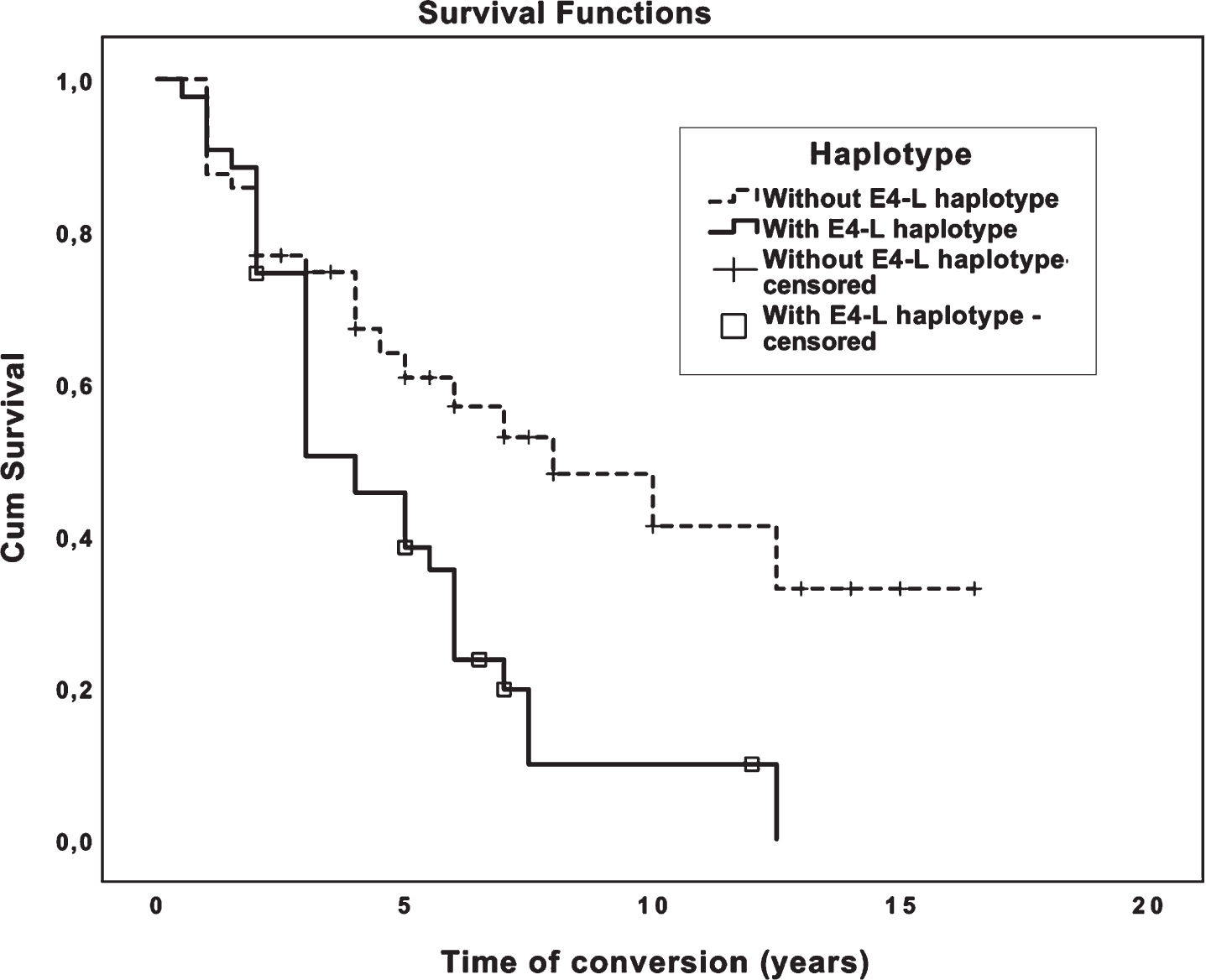
Survival analysis curves (Kaplan-Meier) for mean time of conversion from MCI to AD (y) for patients with and without ɛ4-L haplotype.
APOE ɛ4-TOMM40′ 523 L haplotype impact on AD biomarkers
We further observed that AD biomarkers significantly increased the risk of MCI conversion to AD. Indeed, when classified as having a profile compatible with AD, using the dichotomic variable IATI index [53], AD biomarkers significantly increased the risk of conversion from MCI to AD (OR = 15.6, p < 0.001, 95% CI: 5.2 to 46.3). We also evaluated the association of ɛ4-L haplotype with biomarkers and verified that this haplotype is significantly associated with biomarkers compatible with AD (p = 0.007) (Table 5). Considering the values of the 3 biomarkers: Aβ42, t-Tau, and p-Tau we further observed that the ɛ4-L is significantly associated with lower levels of Aβ42 (p = 0.015) and higher levels of t-Tau (p = 0.006) and p-Tau (p < 0.001) (Fig. 4).
Cross tabulation of ɛ4-L haplotype and biomarkers classified as having a biomarker profile compatible or not compatible with AD (according to IATI index)
More cases than expected are highlighted in grey. ɛ4-L haplotype is associated with a biomarker profile compatible with AD whereas patients without this haplotype are more associated with biomarker profile not compatible with AD.
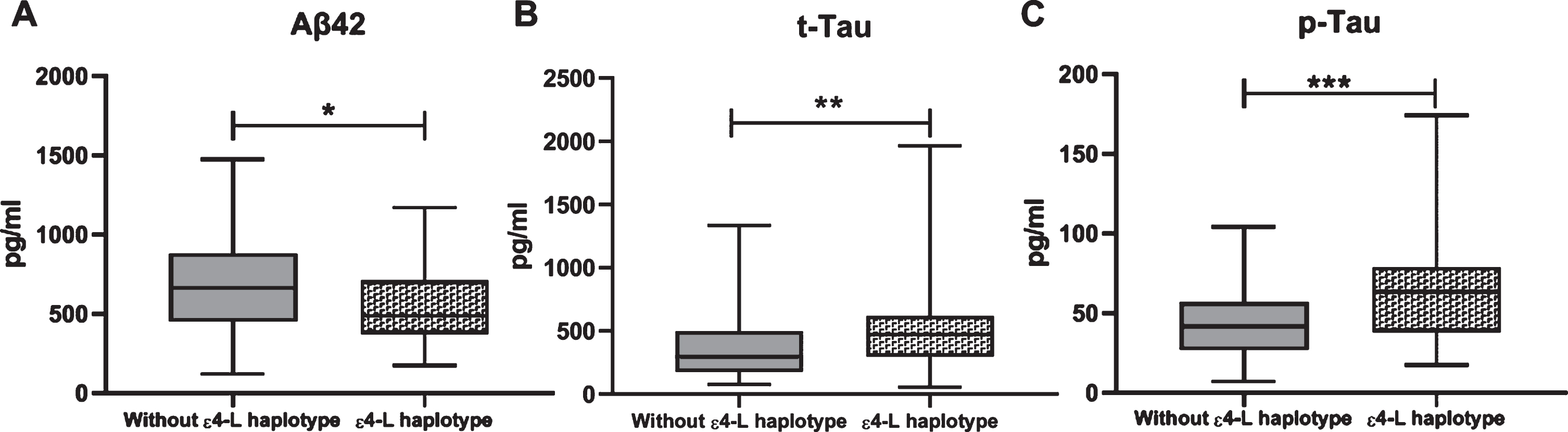
Box plots for CSF concentrations (pg/ml) of Aβ42, t-Tau, and p-Tau (means±SD). Statistical significance between groups was determined using Mann Whitney test *p < 0.05; **p < 0.01; ***p < 0.001.
DISCUSSION
In this study, we uncover the role of the APOE-TOMM40 haplotype on the risk and time of conversion from MCI to AD. We first demonstrated that MCI non-converters (MCI-S) and converters (MCI-AD) had a different poly-T distribution, where the L allele was significantly more frequent in the MCI-AD group. We further evaluated how this difference impacted the risk of conversion and found that, having at least one L allele significantly increased the risk of conversion from MCI to AD. Due to the strong LD between APOE and TOMM40 we were unable to statistically distinguish the independent effects of TOMM40-L and APOE ɛ4. As it was not possible to disentangle if the effect would be due to APOE ɛ4 allele or due to TOMM40′ 523 L allele, we analyzed instead the APOE ɛ4-TOMM40′ 523 L haplotype performing a haplotype analysis and verified a significantly higher risk of conversion for the ɛ4-L haplotype carriers and that patients carrying this haplotype had significantly lower mean times of conversion. Lastly, we observed that the ɛ4-L haplotype was significantly associated with a CSF biomarker profile compatible with AD, namely, significantly lower levels of Aβ42 and higher levels of Tau and p-Tau.
Very few studies have focused on the impact of TOMM40′ 523 polymorphism on MCI and, to our knowledge, none of them studied the impact of APOE-TOMM40′ 523 haplotype on risk and conversion time, as we did in this study. Roses' group, who identified this polymorphism, presented a stratification by TOMM40 ′523 and APOE genotype of ages of onset of cognitive impairment, in order to create a risk algorithm for clinical trial enrichment [22, 23]. The major difference between these two studies and our study is that the other authors evaluated the risk of conversion from normal cognition to MCI and/or AD, whereas in our study the event studied was the risk of conversion from MCI to AD. Another study evaluated the utility of TOMM40 poly-T variable-length polymorphism among other 247 variables, for modelling the progression from MCI to AD but the TOMM40 ′523 genotype variable was not present in the final model [25]. On the other hand, it was not clear how the TOMM40 ′523 poly-T variable was classified. Lastly, Laczo et al. studied the impact of TOMM40 ′523 genotype on cognition and brain structure among aMCI individuals [26]. In contrast, regarding the APOE ɛ4 allele, dozens of studies reviewed in previous metanalysis [7, 8] showed that ɛ4 is a risk factor for the progression from MCI to AD, despite its low sensitivity [10].
CSF biomarkers are also used as predictors for the progression from MCI to AD [11]. Li et al. showed in a metanalysis that abnormal levels of t-Tau, p-Tau, and the ratio t-Tau/Aβ42 are associated with high risk of progression from MCI to AD [8]. Here, we also demonstrated that MCI patients with a biomarker profile compatible with AD, using the dichotomic variable IATI index [53], were significantly at higher risk to convert from MCI to AD, and that ɛ4-L haplotype was significantly associated with a biomarker profile compatible with AD, namely lower levels of Aβ42 and higher levels of t-Tau and p-Tau. Previous studies also demonstrated a similar influence of APOE ɛ4 on CSF AD biomarkers [55–57]. Regarding TOMM40′523 L, the associations observed with this allele and CSF biomarkers were attributed to ɛ4 allele [20, 59]. Taking all this data into consideration, we hypothesize that the higher risk of conversion and lower mean times of conversion observed in ɛ4-L haplotype carriers, can be driven by CSF biomarkers.
In the present study, we analyzed the role of APOE ɛ4-TOMM40′523L haplotype on the risk of MCI to AD conversion instead of APOE ɛ4 and TOMM40′ 523 L allele separately as explained earlier. A recent study also addressed the impact of the APOE-TOMM40 haplotype on susceptibility to dementia with Lewy bodies, Parkinson’s disease dementia, and Parkinson’s disease in clinically and neuropathologically well-characterized individuals and found that the ɛ4-L haplotype increased the susceptibility and risk of earlier dementia with Lewy bodies onset. This association was explained by the co-occurrence of AD pathology [17]. Although in the Caucasian population the ɛ4 allele is almost exclusively associated with L allele, as demonstrated in this study, the same does not occur in other populations such as in Ghanaian, African Americans, and Japanese population where, ɛ4 is commonly linked to ′523-S in addition to the ′523-L allele [48, 61]. Considering this, Yu et al. compared the ɛ4-L haplotypes between older Caucasians and African Americans and demonstrated that the effect size and effect pattern on AD dementia incidence were similar between ɛ4 and ′523-L on the Caucasian, but different in the African carriers. In the same study it was also demonstrated that the risk conferred by the APOE ɛ4 haplotype depends on the TOMM40′523 allele in LD, showing that African Americans with ɛ4-′523-L haplotype show stronger effect on the increased risk for AD dementia than those with either ɛ4-′523-S or ɛ4-′523-VL haplotypes [62]. In line with these results, Prokopenko also suggested the possibility that part of the liability of LOAD, commonly ascribed to ɛ4, might have been caused by TOMM40 on the basis of its strong LD [17]. Thus, we propose the ɛ4-L haplotype as a risk factor for MCI to AD conversion and that its effect may be due to the combination of both APOE and TOMM40 genes. APOE and TOMM40 encode two very distinct proteins. APOE encodes the Apolipoprotein E (ApoE) protein, which is a glycoprotein that regulates lipid homeostasis by mediating lipid transport from one tissue or cell type to another [63, 64]. However, other important roles have been described for this protein, such as Aβ metabolism and clearance, tau phosphorylation, brain activity and atrophy, brain cholesterol transport, synaptic plasticity, inflammation, and brain neurogenesis (reviewed in [64]). On the other hand, TOMM40 encodes the channel subunit of the outer mitochondrial membrane protein complex: TOM40, through which the majority of nuclear-encoded proteins enter mitochondria [65, 66], including Aβ and AβPP, which further lead to mitochondrial dysfunction [67, 68]. Mitochondria dysfunction has been shown to be an early and well characterized event in AD [69, 70]. APOE ɛ4 contributes to mitochondrial dysfunction, decreasing mitochondrial mobility, expression of mitochondrial respiratory enzymes, and translocases of the inner and outer mitochondrial membranes (TIMs and TOMs, respectively) [71]. It was also demonstrated that ApoE4 (1–272), a bioactive carboxyl-terminal-truncated product from ApoE4 proteolytic cleavage [72], is internalized into mitochondria, causing mitochondrial dysfunction [73, 74], by reducing mitochondrial potential [73] and by bounding several mitochondrial proteins such as ubiquinol cytochrome c reductase core protein 2 and subunit 4 of cytochrome oxidase [74]. ApoE4 (1–272) internalization probably occurs via TOM40 [75] which shows an interaction between the two genes of the APOE-TOMM40 haplotype (Fig. 5). To our knowledge only two studies focused on the relation of TOMM40′ 523 and mitochondrial function whose results are contradictory [76, 77]. Therefore, further studies addressing the cellular and mitochondrial role of APOE-TOMM40 haplotypes are needed, namely studying how TOMM40′ 523 alleles can affect ApoE internalization into mitochondria and subsequent mitochondria function.
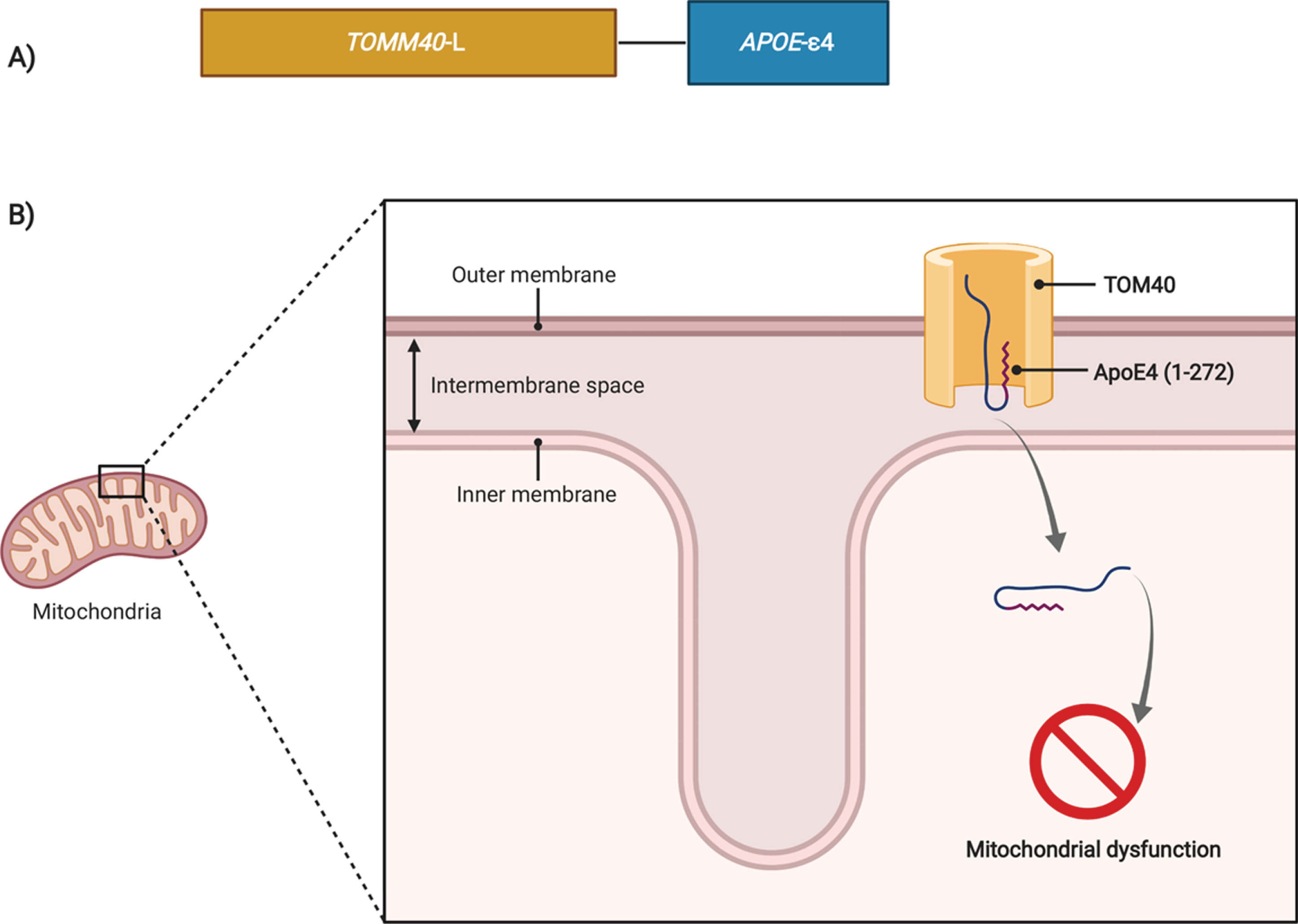
Possible biological interaction between APOE and TOMM40 genes. A) The TOMM40 gene encodes the channel subunit of the outer mitochondrial membrane protein complex TOM40, through which the majority of nuclear-encoded proteins enter mitochondria. On the other hand, APOE codes the Apolipoprotein E (ApoE) protein, which is a glycoprotein that regulates lipid homeostasis by mediating lipid transport from one tissue or cell type to another, nevertheless other important roles in AD pathophysiology have been described for APOE. B) ApoE4 (1–272) a fragment from full ApoE4, was demonstrated to be internalized into mitochondria, causing mitochondrial dysfunction. ApoE4 (1–272) internalization probably occurs via TOM40 channel which shows an interaction between the two genes, which can have a possible modulatory effect, turning the APOE-TOMM40 haplotype analysis important to address this possible joint effect. Therefore, studies addressing how TOMM40′523 alleles can affect ApoE internalization into mitochondria and subsequent mitochondrial dysfunction are also needed.
In our study, two main limitations can be found: the low dimension of the sample of MCI subjects and the lack of familial data. The low number of MCI subjects and conversion events decreased the statistical power when we stratified by TOMM40′ 523 genotypes. The same happened when analyzing the ɛ3/ɛ3 and ɛ3/ɛ4 stratum, thus making it impossible to find any TOMM40 ′523 independent effect. We tried to overcome this limitation with specific statistical analysis focusing on APOE-TOMM40′523 haplotype. Furthermore, in the logistic regression analysis, we tried to adjust for several confounding factors and applied multiple testing corrections. Regarding the lack of familial data, that made it impossible for us to establish the phase of the haplotype with the parent-offspring transmission, because we could not assess if the APOE ɛ4 and the L allele were in cis or in trans.
One of the main strengths of our study is the fact of being performed in a well-characterized cohort of patients both clinically and regarding CSF biomarkers of disease. Furthermore, the data presented here reinforce the hypothesis of mitochondrial dysfunction as an early event in the pathophysiology of AD.
In summary, our study shows that the APOE ɛ4 - TOMM40 ′523 L haplotype is associated with a higher risk and shorter times of conversion from MCI to AD possibly driven by CSF biomarkers and mitochondrial dysfunction.
Footnotes
ACKNOWLEDGMENTS
The authors’ wish to thank all patients and their caregivers who participated in the study. R.C. was funded by the Portuguese Foundation for Science and Technology (FCT) through PhD fellowship SFRH/BD/51995/2012 in the framework of the Inter-University Programme in Ageing and Chronic Disease (PhDOC), a collaboration between the Faculty of Medicine of the University of Coimbra, the NOVA Medical School of the NOVA University of Lisbon and the School of Medicine of the University of Minho and their associated research centres. The work was funded by the European Regional Development Fund (ERDF), through the Centre Regional Operational Programme 2020 under project name/code: CENTRO-01-0145-FEDER-000008: Brain Health 2020, and via the COMPETE 2020 - Operational Programme for Competitiveness and Internationalization and Portuguese national funds via FCT, under project[s] POCI-01-0145-FEDER-007440. We also want to thank to Duarte Barral, PhD for critical reading of the manuscript.