
Abstract
Specific protein misfolding and aggregation are mechanisms underlying various neurodegenerative diseases such as prion disease and Alzheimer’s disease (AD). The misfolded proteins are involved in prions, amyloid-β (Aβ), tau, and α-synuclein disorders; they share common structural, biological, and biochemical characteristics, as well as similar mechanisms of aggregation and self-propagation. Pathological features of AD include the appearance of plaques consisting of deposition of protein Aβ and neurofibrillary tangles formed by the hyperphosphorylated tau protein. Although it is not clear how protein aggregation leads to AD, we are learning that the cellular prion protein (PrPC) plays an important role in the pathogenesis of AD. Herein, we first examined the pathogenesis of prion and AD with a focus on the contribution of PrPC to the development of AD. We analyzed the mechanisms that lead to the formation of a high affinity bond between Aβ oligomers (AβOs) and PrPC. Also, we studied the role of PrPC as an AβO receptor that initiates an AβO-induced signal cascade involving mGluR5, Fyn, Pyk2, and eEF2K linking Aβ and tau pathologies, resulting in the death of neurons in the central nervous system. Finally, we have described how the PrPC-AβOs interaction can be used as a new potential therapeutic target for the treatment of PrPC-dependent AD.
Keywords
INTRODUCTION
Cellular prion protein (PrPC), a molecule discovered by Stanley Prusiner [1], is present in all nucleated cells although it is mainly expressed in neuronal cells [2]. Prion protein has two possible tridimensional (3D) conformations: the physiological isoform PrPC and the scrapie prion protein isoform (PrPSc), which is involved in the transmissible spongiform encephalopathies (TSEs) [3] such as scrapie in sheep, chronic wasting disease in deer, bovine spongiform encephalopathy in cattle, and Creutzfeldt–Jakob disease (CJD), Fatal Familial Insomnia, Gerstmann–Sträussler–Scheinker syndrome in humans [4]. In humans, the mature PrPC protein is encoded by the PRNP gene, located on the short arm of chromosome 20 [5, 6]. PrPC is a highly conserved glycoprotein bound to the cellular membrane by means of glycosylphosphatidylinositol (GPI)-anchor [7]. The protein is associated with lipid raft microdomains [8], sub-compartments of the plasma membrane mainly enriched in cholesterol and glycosphingolipids such as GM3, GM1, and GD3 [9] and proteins involved in signal transduction [10]. Following its biosynthesis, PrPC traffics dynamically through diverse membrane compartments to be processed, glycosylated, properly folded, and then correctly anchored on the plasma membrane [11]. PrPC is formed by a N-terminal region, with 5 octapeptide repeats able to bind the Cu2+ ions, a middle region that contains a cluster of lysine residues and a hydrophobic domain [12], and a C-terminal globular domain which contains 3 α-helices, 2 short β-sheets, and interconnecting loops [13, 14]. Moreover, a disulfide bond is found between residues 179 and 214 [15], and some N-linked glycans can be added at residues 181 and 197 [16]. PrPC and PrPSc have the same molecular weight, the same amino acid and oligosaccharide composition as they are synthesized by the same gene, but they have a distinct 3D conformation: PrPC is enriched with α-helical content (42%), and it has a little β-sheet structure (3%), whereas PrPSc shows less α-helical content (30%) and is rich in β-sheet structure (43%) [17, 18]. Over time, PrPC has been associated with an astounding variety of biological processes including neuronal homeostasis, neuroprotective and pro-myelinating functions, neuronal differentiation process, neurotransmission, stem cell fate, protection against stress or cell adhesion, zinc-copper transport, and calcium homeostasis [19, 20]. The involvement of PrPC in so many activities can be explained by the role of PrPC in cell signaling events [21]. Firstly, the ability of PrPC to bind Cu2+ ions in the fractions of the brain membrane and to improve the incorporation of copper into superoxide dismutase [22], suggest a possible antioxidant activity of the protein [23], that could regulate the influx of Cu2+ into neurons and exerts a protective activity against Cu2+ excess [24]. Indeed, there is a lot of evidence to suggest that Cu2+ excess in cells may be involved in the conformational conversion of PrPC and in the transmission of prion diseases [25]. Furthermore, since PrPC is preferentially localized in the pre- and postsynaptic compartments of nerve endings, it has been thought that it may be involved in preserving normal synaptic structure and function by regulating synaptic transmission and plasticity [26]. Mattei et al. examined the hypothesis that PrPC plays a role in the receptor-mediated apoptotic pathway [27], and in a recent study it has been shown that PrPC may act as an antiapoptotic agent by blocking some of the internal environmental factors that initiate apoptosis [28]. Recent studies documented the involvement of PrPC in angiogenesis, a process in which the increase of PrPC is mediated by hypoxia [29]. Additionally, in vitro studies proposed a role for PrPC in the regulation of neuritogenesis [24, 30], in the axonal growth [31], and in tumorigenesis by regulating tumor growth, differentiation, and resistance to conventional therapies [32, 33]. In the last years, several scientists have highlighted an active role of PrPC in stem cell biology [20, 34]. PrPC is expressed in a wide variety of stem cells such as embryonic and hematopoietic stem cells, taking part in the modulating of proliferation and self-renewal capacity [35], stemness, and in the neuronal differentiation of neural stem cells [36]. Lately, Martellucci et al. demonstrated the presence of PrPC in human dental pulp derived mesenchymal stem cells (hDPSCs) and its role in the neuronal differentiation process [6, 37]. It also demonstrated that the integrity of the lipid raft microdomains is essential for PrPC-induced signaling pathways and that it is essential for hDPSCs’ neuronal differentiation process induced by epidermal growth factor and basic fibroblast growth factor [38].
PrP
C
REFOLDING MECHANISM AND ROLE OF PRIONS IN NEURODEGENERATIVE DISEASES
Mechanism of PrP C refolding
Mammalian prions, the pathogens that cause TSEs, are transmissible particles devoid of nucleic acid composed exclusively of a modified protein that reproduces by recruiting PrPC and stimulating its conversion into infectious isoform PrPSc [39]. Once PrPSc is introduced into individuals from the environment or is generated endogenously, it converts the PrPC into additional molecules of PrPSc [40]. PrPSc propagate by self-perpetuating the structural information stored in the abnormally folded, aggregated conformer PrPSc of the host encoded PrPC [41]. PrPC is converted into PrPSc through specific mechanisms involving a post-translational process during which it acquires a high content of β-sheets [42, 43]. Two distinct mechanisms have been proposed to account for such behavior.
In a first model, the formation of PrPSc is a nucleation-dependent polymerization process. In the absence of a pre-existing aggregate, the conversion between PrPC and PrPSc is reversible, but the PrPSc monomer is less stable than PrPC. The PrPSc aggregates, however, promote the conversion of PrPC by binding and stabilizing the PrPSc conformation.
In a second model, defined “template-assisted mechanism”, the PrPSc form is intrinsically more stable than PrPC, but kinetically inaccessible [3]. In this case, PrPSc could promote conversion by catalyzing the rearrangement of a PrPC molecule, or a partially destabilized intermediate, to the more stable PrPSc conformation and infectivity would therefore be based on the ability of the PrPSc molecule to bind and catalyze the conversion of existing intermediate molecules [44].
Several studies have shown that lipid rafts are important in the refolding process of PrPC in PrPSc [45, 46]. The increasing of the membrane-anchored PrPC local concentration seems to be able to induce a conformational transition accompanied by di- or oligomerization of the PrPC, and that membrane anchoring of an excess of prion protein is the structural prerequisite in the development of prion diseases [9, 47]. Misfolding of the PrPC into the amyloidogenic isoform PrPSc is a key pathogenic event in prion diseases [48] that can present themselves as genetic, infectious, or sporadic diseases. The conformational modification of PrPC in PrPSc with a chain reaction [1], in which PrPSc isoform stimulates the conversion of PrPC in the brain, and the accumulation of these abnormal isoforms, leads to the appearance of highly structured amyloid fibers, which finally form plaques. A similar mechanism of misfolding and aggregation in fibrils and amyloid plaques could be reproduced by a variety of proteins in various diseases such as AD, Parkinson’s disease, amyotrophic lateral sclerosis, frontotemporal dementia, and Huntington’s disease [49].
Cellular and molecular mechanisms of prion neurotoxicity
While a great deal is now known about the mechanisms of prion infectivity and propagation, we have a much more limited understanding about how misfolded PrPSc damages neurons and causes the neuropathological abnormalities characteristic of the disease [40]. Evidence suggests that infectivity (the ability to self-propagate) and neurotoxicity (the ability to produce neuropathology) may be distinct properties attributable to different molecular forms of misfolded PrP [50]. Although recent studies with other amyloidogenic proteins suggest that ordered pre-fibrillar or oligomeric forms may be responsible for cell dysfunction, the precise nature of the neurotoxic species and the mechanism of cell death have yet to be determined. In a study, Sanghera et al. folded the recombinant prion protein (rPrP) into two distinct, β-sheet-rich forms with an intact disulfide bond, noting how the structural properties of the globular and pre-fibril aggregates of rPrP in both states are toxic to neuronal cells in culture [51]. Considerable evidence show that PrPC plays an essential role in mediating prion neurotoxicity, beyond its function as a required precursor to PrPSc [52, 53]. In this regard, it has been hypothesized that PrPC may act as a cell surface receptor that binds PrPSc and transduces downstream neurotoxic signals, a process that could involve the subversion of a normal, physiological activity of PrPC [50, 54]. Furthermore, it is likely that the oligomeric forms of misfolded PrP have been found to be more neurotoxic than large self-propagating PrPSc polymers [55]. An important clue to the mechanism underlying prion neurotoxicity is the observation that PrPC knockout neurons are relatively resistant to the toxic effects of PrPSc that is supplied exogenously by wild-type astrocytes or by neighboring neurons [52, 53]. This result suggests that a critical neurotoxic signal is generated as part of the process by which endogenous cell surface PrPC is converted into PrPSc and, in the absence of PrPC, this signal is not produced. As PrPC is normally attached to the cellular membrane by a GPI-anchor [56], one might predict that a signal-transducing function for PrPC would require its membrane anchoring. Consistent with this prediction, scrapie-inoculated mice expressing an anchorless form of PrPC show an altered neuropathological profile, suggesting that the neurotoxic signaling processes normally mediated by PrPC require its attachment to the plasma membrane [57, 58]. While there are several studies suggesting signal-transducing activities for cell surface PrPC [59], the pathways by which its interaction with PrPSc produces neurotoxic signals remain mysterious. Although the mechanism of neurodegeneration and the involvement of PrPSc is far from clear, data indicates that neuronal apoptosis might be related to activation of several signaling pathways, including proteasome dysfunction, alterations in prion maturation pathway and endoplasmic reticulum (ER) stress. Castilla et al. describe a molecular mechanism of PrPSc neurotoxicity in which the key step in the pathogenesis of prion disorders, regardless of their etiology, is the alteration of ER homeostasis due to drastic modifications of the physico-chemical properties of PrP, which leads activation of ER-dependent signaling pathways that control cell survival [60]. Like other proteins that traffic through the ER, misfolded PrP is retrograde transported to the cytosol for degradation by proteasomes. The accumulation of even small amounts of cytosolic PrP has been found to be strongly neurotoxic both in cultured cells and in transgenic mice: the mice developed normally but acquired severe ataxia, with cerebellar degeneration and gliosis. This establishes a mechanism for converting wild-type PrPC to a highly neurotoxic species that is distinct from the self-propagating PrPSc isoform and suggests a potential common framework for several neurodegenerative disorders [61].
Prions and neurodegenerative diseases
PrPC plays a central role in prion diseases, a set of fatal and incurable neurodegenerative disorders. These disorders share a common molecular mechanism, which is the conformational conversion of the GPI-anchored, properly folded PrPC into the infectious PrPSc that accumulates in the brain of affected individuals [62]. A list of the best-known human TSEs follows:
Kuru
A neurodegenerative, not inflammatory, and infectious disease caused by cannibalism practices. The most typical feature is amyloid “kuru” plaques, which are present in most of cases. Shrunken neurons with dispersed Nissl bodies and intracytoplasmic vacuoles may be present, as well as vacuolated striatal neurons and cerebellar Purkinje cells. A neuropathological feature may be a spongiform transformation and neuronophagy affecting predominantly the deeper cortical layers without involvement of hippocampal neurons. Microglial and astroglial proliferation can also be detected [63, 64].
Creutzfeldt–Jakob Disease
The most common prion disease in humans, CJD is a transmissible and rapidly progressive degenerative disease of the central nervous system (CNS) caused by an accumulation of pathologically conformed PrP. It can be classified into four major phenotypic variants, according to molecular, histopathological, and clinical features: sporadic (sCJD), familial (fCJD), iatrogenic (iCJD), and variant CJD (vCJD). Neuropathological changes include spongiform transformation, neuronal loss, astrocytosis, and the formation of PrP-amyloid plaques in the gray matter, although they occur in only 10–15% of patients with the MV2 subtype of sCJD [65]. Different subtypes of sCJD are distinguishable, to depending on the amino acid specified at the codon 129 Met/Val polymorphic site in PRNP and the type of proteinase K-resistant prion protein fragments, using a western blot examination [66–68].
Structurally, the amyloid plaques of CJD patients show marked heterogeneity. In particular, the plaques present in subjects affected by sCJD are reminiscent of the unicentric stellate plaques of patients with Kuru, being characterized by a dense center of interwoven fibrils and radiating fibrils at the periphery with prevalent absence of dystrophic neurites. Florid plaques, the typical vCJD plaques, are also unicentric but the architecture is less compact and more diffuse, moreover abnormally configured neuronal processes are identifiable in the core and in the edge of the plaque. The form and structure of florid plaques is comparable to the architecture of neuritic plaques, characterized by a central zone of dense amyloid enclosed in a corona of dystrophic Tau-positive neurites [69] commonly observed in the brain parenchyma of AD patients.
Gerstmann–Sträussler–Scheinker syndrome
Slowly progressive hereditary autosomal dominant neurodegenerative disease or encephalo(myelo)pathy with multicentric PrP plaques localized in the cerebral and cerebellar cortex and the basal ganglia [70].
Fatal Familial Insomnia
A rare autosomal-dominant inherited prion disease characterized clinically by severe sleep disorder, motor signs, dysautonomia, and abnormal behavior. Fatal familial insomnia is associated with the aspartic acid to asparagine substitution at codon 178 of the prion protein gene [71, 72].
Lately, it has been discovered that some neurodegenerative diseases such as AD, Parkinson’s disease, amyotrophic lateral sclerosis, frontotemporal dementia, and Huntington’s disease share common pathogenic mechanisms with prion diseases, including the presence of misfolded protein deposits, protein aggregation and progressive neuronal loss in specific areas of the brain. The misfolded proteins involved in these disorders (amyloid-β (Aβ), tau, and α-synuclein) share common structural, biological, and biochemical features, as well as similar mechanisms of aggregation and self-propagation [73]. These deposits composed of Aβ, or α-synuclein protein spread from cell to cell, in a prion-like manner and emerging evidence suggests that the circulating soluble species of these misfolded proteins, the oligomers, could play an important role in the development of pathology while, the less toxic insoluble aggregates would exercise a protective function [74, 75].
Recent evidence suggests that PrPC can act as a toxicity transduction receptor for amyloid-β oligomers (AβOs) and for this reason it is becoming clear that PrPC can play an important role in the pathogenesis of AD [76]. Pathological features associated with neurodegeneration in AD include the formation of plaques due to the deposition of AβOs and neurofibrillary tangles (NFTs) formed by the hyperphosphorylated tau protein [77]. AβOs bind directly to PrPC [78], and it is this AβOs-PrPC interaction that plays an important role in Aβ toxicity by aggravating its interference with synaptic plasticity [79].
ALZHEIMER’S DISEASE: AMYLOID PLAQUES AND NEUROFIBRILLARY TANGLES
AD is a chronic neurodegenerative disease and one of the most common forms of dementia [80]. AD manifests with symptoms such as: short-term memory loss, visual-spatial perception disorders, and impaired language and executive functions [81].
From a neuropathological point of view, AD is characterized by extracellular amyloid plaques and NFTs located within the cells. Although different in shape, density, and localization, these deposits cooperate in destroying CNS regions responsible for learning and memory, the hippocampus, and the neocortex [82]. Amyloid plaques are aggregates of Aβ peptide, a product of natural cellular metabolism, consisting of several amino acids ranging between 36 and 43. The Aβ peptides derive from the proteolytic cleavage of amyloid-β protein precursor (AβPP), a transmembrane protein expressed by the cells of various organs such as the brain, heart, spleen, and kidneys.
NFTs are filamentous inclusions typical of AD and other neurodegenerative diseases called “tauopathies”, located within pyramidal neurons. The number of NFTs found in brain tissue is considered a pathological marker of the dementia severity. The main component of such neuropathological lesions is an aggregated and hyperphosphorylated form of the tau protein. Tau is an abundant axonal soluble protein, and it promotes the microtubule assembly and stability and regulates the molecules and cell organelles transport [83]. The hyperphosphorylated form of tau observed in tauopathies is thought to contribute largely to neuronal degeneration and indirectly, to cell death [84].
Amyloid Precursor Protein (APP) and Alzheimer’s disease
Studies on APP as a genetic determinant in AD began in the mid-1980s observing that individuals with Down syndrome who survived over 30–40 years of age developed the classic neuropathological and clinical characteristics of AD. These data are focused on the involvement of chromosome 21 in AD, supporting the theory that the over-expression of a gene located on this chromosome and present in double copy in Down syndrome, could reproduce the clinical phenotype and neuropathology of subjects with AD. Studies carried out on this pathology were able to identify the first linkage between a locus of chromosome 21q and the familial form of early-onset AD. During the same period, other groups located the gene coding for APP on chromosome 21, which became the first candidate gene responsible for the inheritance of AD. Finally, sequencing and screening for mutations were carried out which unequivocally demonstrated that APP was the gene locus of the disease [85]. The gene product is represented by a ubiquitous type 1 membrane glycoprotein, encoded by the homonymous gene located, as mentioned, on chromosome 21q2.
There are several isoforms produced by alternative gene splicing of 19 exons: exons 1–13, 13a, and 14–18. The predominant transcripts are APP695 (exons 1–6, 9–18), APP751 (exons 1–7, 9–18), and APP770. All these transcripts encode multiple domain proteins with a single intramembrane region. These isoforms differ from each other as in the case of APP751 and APP770 containing 7 exons that encode a serine protease inhibitor domain (Kunitz proteinase inhibitor, KPI). APP695 is the smallest isoform and is the predominant form in neuronal tissue while, APP751 is the predominant form in glial cells. Aβ derives from the region of the protein encoded from its exons 16 and 17 [86]. The nucleotide sequencing of the exon 17 APP gene led to the discovery of numerous missense mutations in families with early onset AD: the first mutation was found in an English family and called the “London mutation”. It is characterized by the substitution of a valine in an isoleucine at codon 717; mutations in codons 670 and 671 were discovered in two Swedish families and consist of a substitution of a base pair: lysine and methionine are replaced by aspartic acid and leucine (“Swedish mutations”) immediately before the N-terminal segment of the peptide Aβ. The “Flemish mutation” at codon 692 (Ala in Gly) causes an intermediate phenotype between cerebral amyloid angiopathy and AD. Other pathogenic missense mutations have been described at codons 716, 715, 714, 694, 693, and 665. Some of these mutations could be responsible for an altered metabolism of AβPP, being at the level of the secretase cleavage site. Two ways of proteolytic processing of AβPP are known: a non-amyloidogenic way and an amyloidogenic way [86]. The non-amyloidogenic pathway involves α-secretase, an enzyme that cuts AβPP, generating a soluble N-terminal fragment (sAβPPα) and a C-terminal fragment anchored to the membrane, CTFα (also known as C83). The amyloidogenic pathway involves β-secretase, an enzyme also known as BACE (β-APP-site cleaving enzyme), which cuts the AβPP at the level of the N-terminal end generating the sAβPPβ fragment and the C- fragment CTFβ terminal (also known as C99). Cleavage of some β-secretases can be displaced by ten amino acid residues, generating the sAβPPβ fragment and the CTFβ ’(or C89) fragment. All C-terminal fragments (C83, C99, and C89) are substrates for γ-secretase, a high molecular weight (>106 kDa) multiprotein enzyme consisting of a presenilin (presenilin 1 or presenilin 2), associated with other components such as nicastrin, anterior pharynx defective 1 (APH-1), and presenilin enhancer 2 (PEN-2). When γ-secretase acts on the C83 fragment determines the formation of the intracellular domain of APP (AICD) and p3 peptide. The action of secretase at the level of the C99 fragment leads to form the AICD and the Aβ1-40 and Aβ1-42 peptides, while on the fragment C89 determines AICD and Glu11Aβ fragments. Both types of Aβ can be found at the level of amyloid plaques, but the Aβ1-42 peptide has a strongly neurotoxic action and a greater tendency to aggregate than the Aβ1-40 form [87]. Under normal conditions, about 90% of the secreted Aβ peptide is Aβ1-40, the soluble form of the peptide that only slowly converts to an insoluble β-sheet configuration and, for this reason, can be readily eliminated from the brain. On the contrary, about 10% of the secreted Aβ peptide is Aβ1-42, which tends to aggregate easily and settle early in the brain in individuals with AD and Down syndrome [84]. At the intracellular level, Aβ is present in the form of monomers, oligomers, protofibrils, and fibrils. While the former does not show pathogenic action, the others can facilitate the hyperphosphorylation of the tau protein, the destruction of the proteasome and mitochondrial functions, the deregulation of calcium homeostasis, the loss of synapses, the decrease in the release of neurotransmitters (acetylcholine) and finally, can lead to the death of neurons [87].
Hypothesis of the amyloid cascade
Research on AD over the past 25 years has been dominated by the hypothesis of the amyloid cascade. According to this hypothesis, a dysfunction of the metabolism of AβPP and the consequent accumulation of Aβ peptides and their aggregation in the form of senile plaques in the brain parenchyma, with consequent neuronal dysfunction and death, represents the crucial event that leads to dementia [88]. In the original hypothesis, these neuronal alterations were attributed by many authors to the toxic effects of the total amyloid. Over the years, knowledge about the pathological features of AD has increased, and this hypothesis was changed, as it became clear how the correlation between dementia or other cognitive alterations and the accumulation of Aβ in the brain as amyloid plaques, were not linear. Research has focused on more specific alterations of Aβ processing, such as the cleavage of AβPP into different peptides, Aβ1-40 and Aβ1-42 and the importance of AβOs (small aggregates of 2 to 12 and more peptides). Several studies have reported that Aβ1-42 aggregates faster than Aβ1-40, instead oligomers have been shown to be more toxic than mature fibrils. Then, the concept of soluble toxic oligomers has been proposed to be responsible for the neurotoxicity of the Aβ peptide. An example comes from Aβ56, which is negatively associated with cognitive decline in an APP mouse model, and some authors have seen that it induces memory deficits when injected into rat brains [89]. These intermediate forms lie between free or soluble Aβ monomers and insoluble amyloid fibrils, but the exact molecular composition of these oligomers has not been fully understood. The hypothesis of the amyloid cascade suggests that synaptotoxicity and neurotoxicity could be mediated by these soluble forms of the multimeric species of the Aβ peptide, whose deposition in the brain parenchyma would represent a crucial phase in the process leading to AD. The dynamics of these species and the poorly defined mechanisms of toxicity make this topic particularly controversial in this field [90]. One of the sticking points of the amyloid cascade hypothesis is that apparently healthy people can have many plaques. These subjects have a lower oligomer-to-plaque ratio than patients with dementia, but it is hypothesized that the plaques sequester soluble oligomers until a limit is reached beyond which the excess oligomers begin to diffuse along the synaptic membranes [91]. Another critical point is that to date no drug targeting Aβ has been recognized to be as effective as disease-modifying. In fact, several possible drugs targeting Aβ have failed to demonstrate efficacy clinical trials [92, 93], even if this might be due to a too late administration of these drugs to reverse the damage. On June 7, 2021, the US Food and Drug Administration (FDA) approved the drug aducanumab, a monoclonal antibody targeted against Aβ for the treatment of AD, however, evidence of clinical benefit from two clinical trials of Phase III is contradictory and inconclusive [94].
Other hypotheses of AD pathogenesis
Among amyloid hypothesis, other pathogenic mechanisms are proposed, first of which is the tau hyperphosphorylation hypothesis. Hyperphosphorylated tau in AD patients’ brains could contribute largely to neuronal degeneration and indirectly, to cell death, through two mechanisms: 1) the removal of tau from microtubules and the consequent destruction of cell trafficking (events leading to dysfunction and loss of synapses); 2) the production of a more fibrillogenic form of tau protein, capable of blocking transport processes and causing cell death. Moreover, an association between the degree of tau aggregation and the pathological severity of AD was described. Further, oxidative stress and inflammation are involved in AD brain pathology. Lipid peroxidation was found in membranes, and other damage stress in proteins and nucleic acids, while inflammatory processes include astrocytes and microglia activation, with higher proinflammatory cytokines [95].
ROLE OF PRION PROTEIN IN ALZHEIMER’S DISEASE
Cellular AβOs receptors a bridge between PrP and amyloid hypothesis
Even after many years of intensive research, the mechanisms that govern the etiology of AD are still partially unknown, also because of the numerous molecular pathways linked to the neurodegenerative process. Proteolytic pro-amyloidogenic processing of AβPP, as mentioned above, triggers the generation of monomeric Aβ peptides, intermediary Aβ oligomers, and high molecular weight β-stranded fibrils that contribute to the development of dementia at several levels [96, 97]. In particular, the heterogeneity of structure, morphology, conformation, and post-translational modification of the soluble amyloid species greatly complicates a mechanistic reconstruction of the AβOs effect on CNS cells [98, 99]. However, accumulating data suggest that Aβ impairment of synaptic plasticity and memory is closely related to biological activity of soluble nonfibrillary Aβ peptide aggregates [100]. Indeed, AβO burden in the brain correlates with synaptic impairment and cognitive decline both in AD subjects [101] and in animal models of the pathology [102]. Further, individuals expressing Osaka mutation of the APP gene develop dementia without the presence of amyloid plaques but accumulate AβOs within neurons [103]. Among the different soluble species of AβO, that can be antigenically distinct from monomeric Aβ peptides [104], some are characterized by a significant ligand-like specificity to cultured hippocampal neurons [105] or brain slice preparations [106] suggesting the presence of cell surface receptors that mediate signal transduction of synaptic selective toxicity. To date, at least 20 possible putative cognate-ligand receptors have been so far proposed [107], but among all the PrPC, showing the highest-affinity for AβOs, can be assumed to have greater relevance to the progressive nature of AD [108]. Several lines of evidence support this assertion: AβOs, monomers or fibrils not included, have been shown to bind directly to PrPC in a cell-based screen with affinity in a nanomolar range and antibodies against PrPC strongly block this interaction [109]. Moreover, spatial learning and memory deficits in animal models of AD depend on the brain expression of PrPC [110] while, antibodies against the PrPc receptor rescue the loss of cognitive performance [111]. Nonetheless, several authors, reported that in certain contexts the pathological effect exerted by AβOs on synaptic dysfunction, reduction of spine density, inhibition of long-term potentiation, and enhancement of long-term depression (both the last two processes are considered models of synaptic plasticity) are independent on the actual presence of PrPc receptors [112]. Most likely, these differences regarding AβOs-induced degenerative cascades are due to the possible interaction of specific species of oligomers with other cell surface receptors able to activate several toxic pathways for synapses and finally neurons. Intriguingly, in a similar way as described for AβOs, membrane PrPC, seems to participate in the establishment of several membrane complexes with numerous β-sheet-rich conformers retaining a prominent role as neurotoxic effector [113].
PrP-dependent downregulation of Fyn signaling cascade from AβO to tau toxicity
Allegedly, the aforementioned physical assembly of the oligomer-PrPC complex remains structurally localized in the external leaflet of the outer surface of the plasma membrane, in particular inside the lipid rafts [114]. The signaling cascade induced by AβOs-PrPC binding and its downstream interacting partners began to emerge with the identification of a close functional relationship of the complex with Fyn [115] (Fig. 1), an intracellular non-receptor tyrosine kinase highly expressed in neurons [116] that belongs to the SRC family kinases. The Fyn kinase plays a role in many biological processes in the CNS, including myelination, oligodendrocyte differentiation, axon outgrowth, long-term potentiation (LTP), and regulation of N-methyl-D-aspartate receptors (NMDARs)[117, 118]. Notably, Fyn overexpression amplifies [119], whereas ablation of the enzyme alleviates memory impairment in AD animal models [120] suggesting a direct involvement in the neurodegenerative mechanisms of the human dementia. Fyn is normally localized in lipid rafts [121] as is PrPC and its activation, depending on the assembly of AβOs (dimers and trimers, not monomers or Aβ*56). PrPC complex can be abrogated by antibodies directed against PrPC [122] or triggered by brain extracts derived from AD patients [115]. Considering the main pathological characteristics of AD, several downstream functional effects exerted by the activation of Fyn kinase can be considered particularly interesting with respect to neurodegeneration and synaptotoxicity. Firstly, Fyn could represent a mechanistic link between the AβOs toxic role in AD and tau pathologies [122]. The physical interaction between the enzyme and the microtubule-associated protein tau leads to tyrosine phosphorylation at the amino terminus [123]. Importantly, antibodies against the prion protein suppress Fyn-dependent tau hyperphosphorylation. Moreover, Fyn and other members of SRC kinases can upstream regulate the activity of Proline-rich tyrosine kinase2 (PyK2), a member of the focal adhesion kinase family, which, in its turn, phosphorylates tau tyrosine residues [124]. Interestingly, PyK2, which is present in neurofibrillary tangles [125] is encoded by the PTK2B, a late-onset AD (LOAD) risk gene identified by a genome wide association study [126]. PyK2 is also a regulator of glycogen synthase kinase-3β (GSK3β), a serine/threonine kinase with an important role in tau hyperphosphorylation and microtubule organization. However, it is not clear if AβOs-induced signaling cascade is directly involved in GSK3β activation [127]. Phosphorylation of tau protein has considerable implications for Fyn’s activity, too. In fact, Mondragon-Rodriguez et al. demonstrated that phosphorylated tau sequesters Fyn kinase in dendrites decreasing its concentration at synapses and generating a drop down of the NMDAR density [128]. Intriguingly, AβOs interaction with PrPC on neuron plasma membrane induces a signaling cascade that links Fyn activation to the NMDAR subunit NR2B phosphorylation [115, 122]. NR2B phosphorylation is essential to stabilize the interaction of NMDARs with the scaffolding protein postsynaptic density protein 95 (PSD95) in order to get a strong binding to the postsynaptic density (PSD) of the receptors [129]. The final effect of the signaling pathway is the inhibition of NMDAR endocytosis that preserves the NMDAR mediated Ca2+ currents [130]. Hyperphosphorylated tau by binding to Fyn in dendrites perturbs this signal cascade disrupting calcium homeostasis within the post-synaptic sites. The binding of AβOs with the PrPC in addition to altering the mobilization of calcium, causes the activation of the eukaryotic elongation factor (eEF2) kinase (also known as calmodulin-dependent protein kinase type III) [131] and the consequent phosphorylation of the eEF2 associated to Aβ-induced LTP failure [132]. The eEF2 factor induces an increased expression of calcium/calmodulin-dependent protein kinase II [133] a serine/threonine protein kinase that regulates neurotransmission and synaptic plasticity. Both, the Fyn and eEF2 kinases bind to the Gαq-protein coupled metabotropic Glutamate Receptor 5 (mGluR5), a key element in signal transduction between the AβO-PrPC complex, anchored by the GPI on the outer side of the plasma membrane and the downstream activation of kinases in the cytoplasm [131]. PrPC and Fyn physically interact with mGluR5 to form a protein complex localized in the postsynaptic densities but there is not a direct binding between AβOs and mGluR5. However, in cortical neurons, there are evidence that AβOs dissociate the interaction of the Homer scaffolding proteins with mGluR5 exclusively if the PrPC is expressed by the cells [131, 133] suggesting that AβO-PrPC complex perturbs the ligand binding profile of mGluR5. Further characterization of mGluR5 role in signaling pathway triggered by exposition to AβOs revealed that the specific antagonists of mGluR5 activity, 2-methyl-6-(phenylethynyl) pyridine (MPEP) and 3-[(2-methyl-1,3-thi-azol-4-yl) ethynyl] pyridine (MTEP), can inhibit Fyn and eEF2 activation in vitro and rescue deficits in learning and memory of AD transgenic models [131]. Another potential track to discern the role played by mGluR5 in AβO-mediated neurotoxicity could be represented by its capacity to physically bind to the NR2 subunit of NMDARs by means of Shank, Homer and PSD95 proteins [134]. At the PSD, the colocalization of ionotropic and metabotropic glutamate receptors suggests a downstream functional mechanism that could be impaired by AβO-PrPC complex interaction; however, the molecular aspects of this hypothesis remain so far elusive. Taken together, this evidence suggests that a well-structured cell signaling pathway, comprising PrPC that serves as membrane receptor, mGluR5 as trans-membrane jointing element and several cytoplasmic kinases as downstream functional effectors, may mediate the neuro/synaptotoxic effect of AβOs. Every one of the elements involved in the signal transduction cascade could represent a main pharmacological target to prevent some of the more dangerous clinical symptoms of AD and indeed some attempts to develop therapeutic strategies are being evaluated.
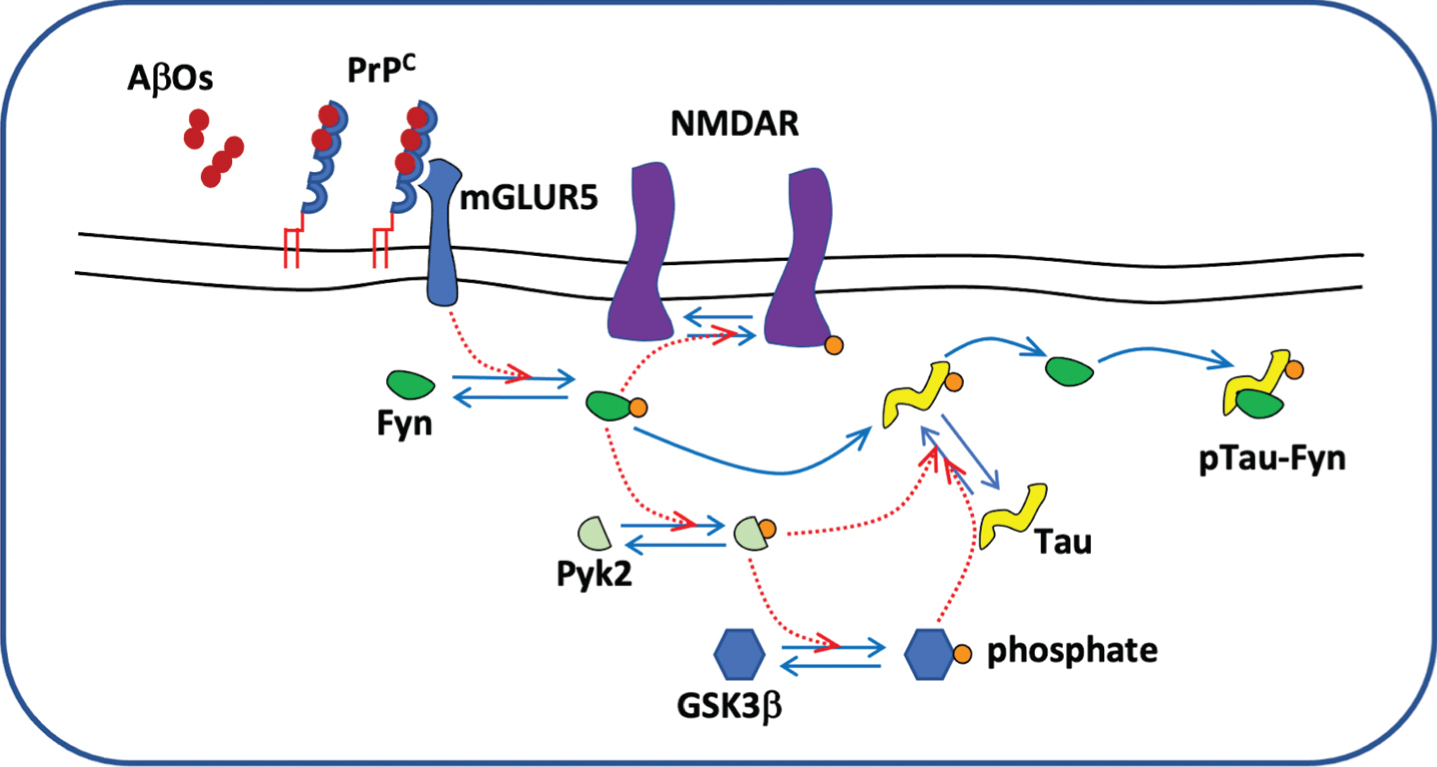
Tau phosphorylation cascade.
Some phase I/II/III clinical trials designed for the treatment of AD
Interaction PrPC- AβOs as new drug target for AD
There is great interest in therapeutic targeting of the toxic signals mediated by AβOs-PrPC complex to intracellular components of the pathway. An early possible therapeutic strategy could be achieved by decreasing the number of oligomers able to bind to the PrPC receptor on the surface of neurons. Indeed, the use of small synthetic peptide inhibitors can stabilize the monomeric forms of amyloid fragments reducing toxic effect of aggregated species [135]. A subsequent therapeutic approach could concern the blocking of the interaction between AβOs and PrPC. Several preclinical data from AD transgenic models demonstrate that antibodies against the amino-terminal residues of PrPC (residues 93–109) prevent binding of AβOs [136, 137] preserving activity of synapses and blocking cognitive decay. Similarly, the small molecule Chicago Sky Blue 6B was able to avert in vitro binding of AβOs to prion protein [138]. However, there are not reports that have investigated therapeutic effects of the drug. Certain investigations show that the soluble PrPC, its N-terminal cleavage fragments [139, 140] or decoy PrPC peptides [141] inhibit disruptive effects on synaptic functions of AβOs reducing the bulk of ligand for membrane anchored PrPC. Leaving further along the amyloid-dependent toxic pathway, another potential therapeutic target could be represented by the physical interaction between the AβOs-PrPC complex and the co-receptor mGluR5. From this perspective Haas et al. evaluated the drug BMS-984923 [142], a potent mGluR5 silent allosteric modulator, showing in an AD transgenic mouse model that the compound hampers pathological AβOs signaling without affecting physiological glutamate transduction cascade [143]. An open-label, single-ascending dose study [144] was started in 2021 to evaluate safety, tolerability, pharmacokinetics, and receptor occupancy of the drug, it is ongoing now. AβO-dependent Fyn kinase activation has been considered another promising pharmaceutical target [145] and some reversible enzyme inhibitors have passed the preclinical development to arrive at clinical research. Saracatinib (AZD0530) is a Src and Abl kinases inhibitor specific for Fyn and Src kinase that was synthesized from anilino quinazoline to arrest the spread of cancer [146]. The drug was repurposed to be tested as AD modifying therapy on the base of its capacity to modulate Fyn activation and protect synaptic densities and cognitive functions in an animal AD model [120, 147]. A phase IB randomized placebo-controlled trial [148] successfully assessed the effect of the Fyn kinase activity inhibition on the primary endpoints concerning safety, tolerability, and compliance to pharmacokinetic parameters in a small cohort of probable AD subjects but fail to demonstrate regional cerebral glucose metabolism preservation as measured by fluorine-18 fluorodeoxyglucose positron emission tomography or clinical efficacy [149]. A subsequent Phase IIA randomized placebo-controlled clinical study [150] enrolled a more consistent cohort of participants diagnosed with mild AD [151]. The conclusion of the second study confirmed safety and tolerability of Saracatinib in subjects affected by mild AD, however highlighted the lack of significant outcomes produced by Saracatinib on both the surrogate endpoint (regional brain glucose metabolism) and cognitive decline or disease biomarker measures. The phenylaminothiazole-type tyrosine kinase inhibitor Masitinib (AB1010) being able to target c-Kit and Fyn enzymatic activity [152] in preclinical models was evaluated as neuroprotective agent in mild to moderate AD by a two double-blind, randomized, placebo-controlled, clinical trial. A first phase II study [153], where Masitinib was administered to AD patients in combination with standard therapies, met its efficacy and tolerability-related outcomes significantly. The add-on therapy was able to reduce the rate of cognitive decline in the treated group respect to the placebo [154]. A following phase IIB/III multicenter clinical trial [155] started in 2013 and seven years after in December 2020 its sponsor, AB Science a late-clinical-stage French company, announced some positive results with respect to clinical efficacy, as measured by the neurological Alzheimer’s Disease Assessment Scale-Cognitive Subscale test and safety. Some concerns, however, have been raised about the number of adverse events recorded during the study that emerged to be almost three times greater in patients treated with Masitinib [156]. Recently, Abd-Elrahman et al. described sex specific difference in PrPC-dependent interaction between AβOs and mGluR5 in an AD animal model [157]. According to the study, the AβOs-PrPC complex binds the glutamate receptor exclusively in male animals, as a consequence the functional effects on cognitive impairment and the underlying Aβ pathology depending on the inhibition of the pathological signal pathway are exclusive to male mice. If confirmed in the humans, these finding would lead to important implications for the design of clinical studies and would suggest the very importance of stratifying by gender the clinical trial outcomes.
CONCLUSION
In this review, we summarize the latest knowledge about the role of PrPC on AD. Indeed, the mechanisms that govern the etiology of AD are still not completely known, partly due to the numerous molecular pathways linked to the neurodegenerative process. Several authors showed that Aβ oligomers but not Aβ monomers are able to bind to PrPC with high affinity and mediate a signal pathway.
The signaling cascade induced by AβOs-PrPC binding and its downstream interacting partners began to emerge with the identification of a close functional relationship of the complex with Fyn, a SRC family kinases. There is great interest in the therapeutic targeting of the toxic signal mediated by AβOs-PrPC complex to intracellular component of the pathway. In this review, we explored different possible therapeutic strategy about the toxic signal mediated by AβOs-PrPC. One of them aims to decrease the number of oligomers able to bind to the PrPC and the possibility to block the interaction between AβOs-PrPC. Several papers showed that the use of antibodies against amino-terminal PrPC or soluble cellular prion protein could prevent the binding of AβOs preserving activity of synapses and blocking cognitive decay. Another possible strategy could be represented by a drug named BMS-984923, a silent allosteric modulator of mGluR5 (a coreceptor of AβOs-PrPC) that is under clinical trials started in 2021 is ongoing now. A Src inhibitor represent a possible strategy to block the downstream of Fyn. The clinical trials on Sarcatinib (IIA phase) showed safety a tolerability of the drug in subject affected by mild AD but highlighted the lack of significant outcomes on both the surrogate endpoint and cognitive decline. A phase IIB/III study on Masitinib (drug able to target c-Kit and Fyn enzymatic activity) showed some positive results respect to clinical efficacy even if some concerns have been raised about adverse events. Despite advances in knowledge of cellular and molecular mechanisms of AD, we are still unable to block or slow down the pathological effects of the disease. We believe that further studies related to possible signal pathways mediated by the interaction between AβOs-PrPC need to be implemented.