
Abstract
Background:
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) caused by NOTCH3 mutations is the most common monogenic hereditary pattern of cerebral small vessel disease. The aggregation of the mutant NOTCH3 may play a cytotoxic role in CADASIL. However, the main mechanism of this process remains unclear.
Objective:
We aimed to investigate the possible pathogenesis of the mutant NOTCH3 in CADASIL.
Methods:
The clinical information of two pedigrees were collected and analyzed. Furthermore, we constructed cell lines corresponding to this mutation in vitro. The degradation of the extracellular domain of NOTCH3 (NOTCH3ECD) was analyzed by Cycloheximide Pulse-Chase Experiment. Flow cytometry and cell counting kit-8 assay were performed to observe the effects of the NOTCH3 mutation on mitochondrial function and apoptosis.
Results:
We confirmed a de novo heterozygous missense NOTCH3 mutation (c.1690G > A, p. A564T) in two pedigrees. In vitro, the NOTCH3ECD aggregation of A564T mutant may be related to their more difficult to degrade. The mitochondrial membrane potential was attenuated, and cell viability was significant decreased in NOTCH3ECD A564T group. Interestingly, BAX and cytochrome c were significantly increased, which are closely related to the mitochondrial-mediated pathway to apoptosis.
Conclusion:
In our study, the aggregation of NOTCH3ECD A564T mutation may be associated with more difficult degradation of the mutant, and the aggregation may produce toxic effects to induce apoptosis through the mitochondrial-mediated pathway. Therefore, we speculated that mitochondrial dysfunction may hopefully become a new breakthrough point to explain the pathogenesis of cysteine-sparing NOTCH3 mutations.
INTRODUCTION
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is the most common monogenic hereditary pattern of cerebral small vessel disease. It results from NOTCH3 mutations on the 19p13 chromosome and is the most important genetic cause of stroke in adults and dementia [1, 2]. The NOTCH3 mutations and granular osmiophilic material (GOM) deposits observed by skin biopsies are regarded as the gold standards for diagnosing CADASIL. This disease usually develops in the middle age, with a slow onset and progressive aggravation. The typical clinical symptoms are migraines with aura occurring in early adulthood, psychiatric disturbances, recurrent ischemic events secondary to lacunar lesions, progressive cognitive impairment with initial presentation of executive dysfunction and motor disabilities, or even dementia [3]. However, the changes are observed on magnetic resonance imaging (MRI) 10–15 years before the onset of clinical symptoms, including white matter hyperintensity (WMH) in the external capsules and the anterior temporal pole, recent small subcortical infarcts, lacunar infarcts (LI) and enlarged perivascular spaces. Progressive WMH and recurrent subcortical infarction are the main causes of cognitive impairment and eventually dementia [2, 4]. Currently, the detailed pathological mechanism of WMH in CADASIL has not been comprehensively elucidated, which may be related to vascular dysfunction caused by GOM deposits [3].
NOTCH3 gene codes a transmembrane protein that is mainly expressed in the vascular smooth muscle cells (VSMC) and pericytes, which are important parts of the blood-brain barrier [5]. This transmembrane protein is composed of an extracellular domain (ECD), a transmembrane domain, and an intracellular domain. The extracellular domain of NOTCH3 protein (NOTCH3ECD) is made up of 34 epidermal growth factor repeats (EGFr) and three LIN-12/notch repeats (LNRs). Each EGFr is composed of approximately 40 amino acids containing six cysteines, which form three disulfide bonds in pairs stabilizing the secondary structure of EGFr. Currently, the classical CADASIL is deemed to be related to NOTCH3 mutations resulting in the loss or acquisition of a cysteine residue in one of the 34 EGFr domains, which disrupts the normal formations of bonds and lead to the misfolding of EGFrs and tertiary structural change in NOTCH3ECD, increasing the aggregation of NOTCH3ECD [6]. The aggregation of NOTCH3ECD outside the cells further recruits other extracellular matrixes to form GOM, causing degeneration of VSMCs and arteriole lesions [7]. Previous studies show that NOTCH3 mutations lead to increased expression of NOTCH3 in VSMCs of CADASIL patients. They also result in alterations in Ca2+ transients and augmented oxidative and endoplasmic reticulum stress responses induced by the increase in nicotinamide adenine dinucleotide phosphate oxidase 5 (NOX5)-derived reactive oxygen species (ROS) levels, thereby, affecting VSMC growth and cytoskeletal reorganization, eventually, causing vascular dysfunction in CADASIL patients [8]. Nicotinamide adenine dinucleotide phosphate (NADPH), a mitochondrial complex, is a major source of mitochondrial ROS [9]. Moreover, mitochondrial dysfunction may result in an imbalance in Ca2+ homeostasis [10]. In addition, the activities of nicotinamide adenine dinucleotide (NADH) dehydrogenase and cytochrome c oxidase in CADASIL patients reduce significantly, and the mitochondrial oxidative phosphorylation chain is damaged [11], further leading to an increase in ROS production [8]. The mitochondrial membrane potential (ΔΨm) decreased significantly along with an increase in the irregular and abnormal mitochondrial VSMCs in three CADASIL patients with R133C mutation [12]. Although these data reflect the effects of only a few NOTCH3 gene mutations, these studies collectively indicate that mitochondrial dysfunction may be involved in the pathogenic mechanism of CADASIL. However, the specific mechanism underlying mitochondrial dysfunction in CADASIL remains unclear.
The first case of CADASIL was reported in 1993 [13]. The prevalence has been predicted to be between 4 and 15 per 100,000 in CADASIL based on epidemiological studies [14]. Currently, more than 300 NOTCH3 mutations are reported worldwide, and most pathogenic mutations located in exons 2–23 of NOTCH3 are related to changes in cysteine. Furthermore, more than 40 cysteine-sparing NOTCH3 mutations were reported in the Human Gene Mutation Database (HGMD), such as G73A, D80G, and L1515P [15 –17]. However, the pathogenic mechanism of cysteine-sparing NOTCH3 mutations remains controversial. In addition, it is not clear whether cysteine-sparing NOTCH3 mutations influence the degradation of NOTCH3ECD, deposits of GOM and cellular functions. In this study, we analyzed the clinical characteristics of two families carrying the same de novo mutation.To evaluate the possible pathogenic mechanism of the novel mutation, we conducted experiments in vitro to observe the effects of the mutation on aggregation of NOTCH3ECD, cell viability, mitochondrial function and apoptosis.
MATERIALS AND METHODS
Clinical assessment of participants and genetic analysis
Two unrelated probands from different families in China were suspected to with CADASIL disease (genealogical tree presented in Fig. 1a). Informed consent was obtained and peripheral venous blood samples from the probands and their family members were collected for NOTCH3 mutation analysis. A total of 120 healthy participants were treated as controls. Written informed consent forms were provided by all participants before being included in the study. The study was authorized by the Ethics Committee of the Henan Provincial People’s Hospital. The new mutation was affirmed using Sanger sequencing and checked with online databases such as gnomAD (https://gnomad.broadinstitute.org), ExAC (http://exac.hms.harvard.edu/), and HGMD (http://www.hgmd.org/). The skin of the left upper limb of two probands were cut to a depth of about 1.5 cm×1 cm and fixed in 2.5% glutaraldehyde phosphate buffer solution. The fixed skin tissue specimens were sent to the Department of Pathology (Xiangya Hospital, Central South University) for GOM detection.
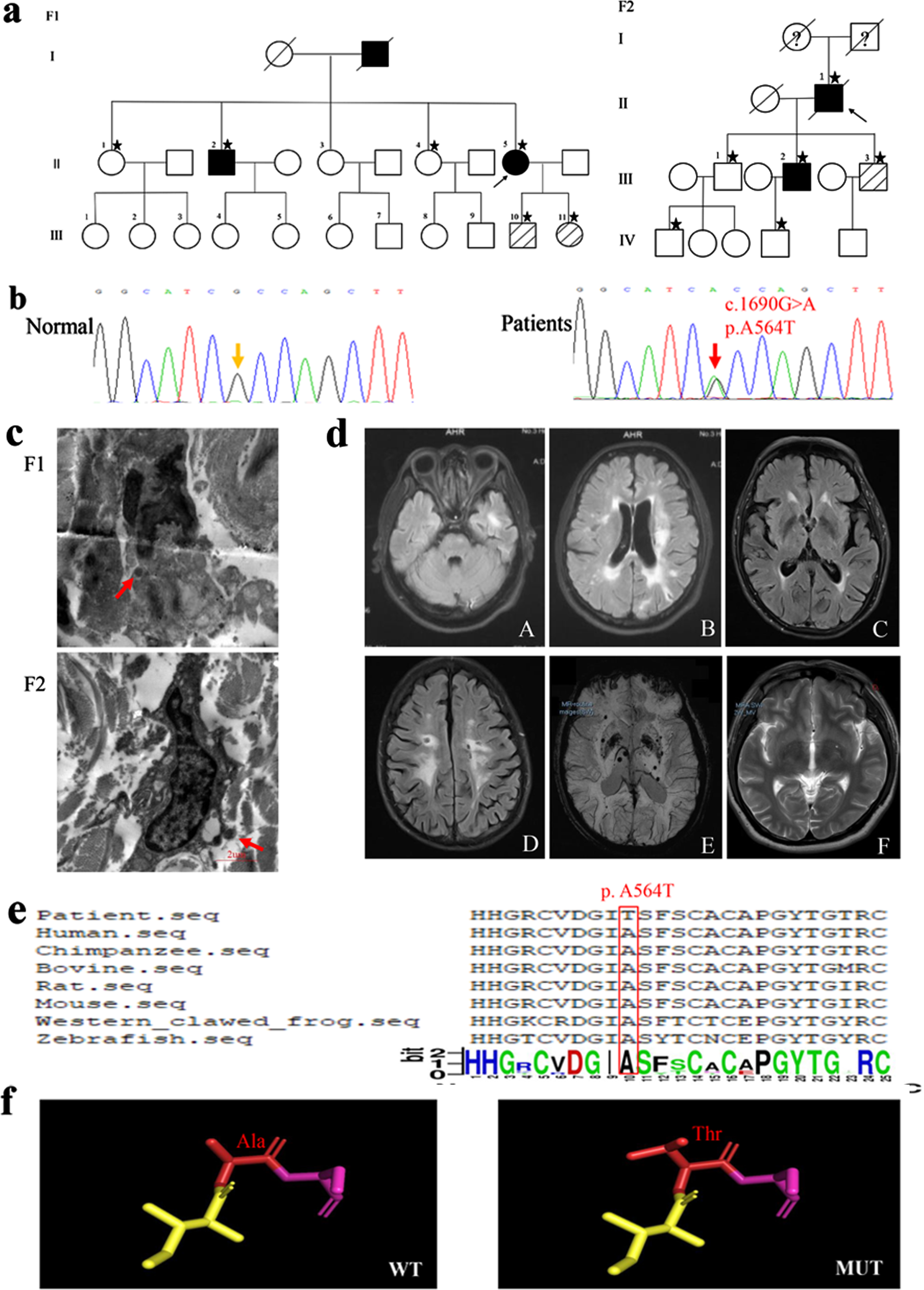
Clinical assessment of participants and bioinformatic analysis. a) Pedigrees of two families. The square represents a man; the circle represents a woman. The arrow refers to the proband. Clinically affected family members are represented in black. Unknown clinical status is indicated by a question mark. Deceased members are represented by single diagonal lines. Gene-tested participants are marked with an asterisk. The three diagonal lines indicate carriers. b) Gene sequencing diagrams. Wild type (Normal) and c.1690G > A (p. A564T) mutation (Patients). c) Representative images of skin biopsies of two probands. GOM deposits are revealed under electron microscopy (red arrows). d) Representative MRI in affected family members. A-B), white matter lesion involving the temporal pole and LI in proband of F1; C-E), white matter lesions involving external capsule, LI and magnetic sensitivity weighted imaging showing microhemorrhage in proband of F2; F), perivascular space in basal ganglia in III.2 of F2. e) The Alanine(Ala) at position 564 in NOTCH3 (red square) is highly conservative among Human (NP_000426.2), Chimpanzee (XP_003316212.1), Bovine (XP_003586294.1), Rat (NP_064472.2), Mouse (NP_032742.1), Western clawed frog (NP_001011355.2), and Zebrafish (NP_571624.2). f) 3D model of NOTCH3 is built from Ala to Thr at position 564 in NOTCH3. WT: wild type, MUT: p. A564T mutation.
We used the algorithms of VarCards (http://varcards.biols.ac.cn/), Mutation Taster (http://www.mutationtaster.org/ChrPos.html), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), and Combined Annotation Dependent Depletion (https://cadd.gs.washington.edu/) to prefigure whether this mutation effected NOTCH3 function. Subsequently, the homologous amino acid sequences of this mutation and other species were retrieved using Uniprot (https://www.uniprot.org/). DNAMAN software and WebLogo (https://weblogo.berkeley.edu/logo.cgi) were used to analyze evolutionary conservation of the amino acids by aligning sequences of NOTCH3 orthologues of multiple species. Furthermore, we used SWISS-MODEL (https://swissmodel.expasy.org/) to construct pre-mutant and post-mutant protein models, whereas the models of wild type (WT) and mutant were simulated by the PyMol software.
Stably transduced cell lines
Human embryonic kidney (HEK) 293T cells (Shanghai, China) were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone, Logan, UT) added 10% fetal bovine serum (FBS) (Gibco) at 37°C in 5% CO2. The lentiviral vectors expressing WT and A564T mutant of NOTCH3ECD and a void vector were purchased from BrainVTA Co., LTD (Wuhan, China). A total of 3×104 293T cells/well were prepared in a 24-wells plate. The following day, the cells in each well were transduced with lentivirus at a multiplicity of infection (MOI) of 6. After 24 h, the transduction media was removed and replaced with fresh DMEM added 10% FBS. Subsequently, the transduced cells were selected using the antibiotic puromycin (2μg/mL) (Sigma, USA) and passaged once every three days in a ratio of 1:3 (Supplementary Figure 1).
Mitochondrial protein extraction
The cells (5×106) harvested were centrifuged at 500 x g for 10 min at 4°C and washed using 0.9% sodium chloride solution. The cell pellet was resuspended with 1 mL ice-cold Lysis Buffer added the Protease Inhibitor Solution (Qiagen, Germany) and hatched for 10 min at 4°C in an end-over-end shaker. After centrifugating at 1000 x g at 4°C for 10 min, the cell pellet resuspended in 1.5 mL of ice-cold Disruption Buffer added the Protease Inhibitor Solution (Qiagen, Germany). The cells were mechanically disrupted using a syringe. After centrifugating again at 1000 x g at 4°C for 10 min, the supernatant collected was centrifuged at 6000 x g for 10 min at 4°C. The pellet contained the mitochondrial fraction and was resuspended in a Disruption Buffer containing the Protease Inhibitor Solutionand. Quantitatively analysis was performed.
Western blot
Cells were harvested and lysed in immunol precipitation (IP) lysis buffer (Beyotime, Shanghai, China), and an ethylenediaminetetraacetic acid (EDTA)-free complete protease inhibitor cocktail tablet (Thermo, USA) was added to extract the total proteins. The mitochondrial proteins were obtained using the Qproteome Mitochondria Isolation Kit (Qiagen, Germany) as described above. Proteins were quantitatively analyzed by the PierceTM BCA Protein Assay Kit (Thermo, USA) and separated through sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes (Millipo, USA). The membranes were blocked in 5% non-fat milk for 1 h and hatched overnight at 4°C with primary antibodies including anti-NOTCH3 mouse monoclonal antibodies (1:1000; Novus, USA), anti-cleaved caspase-3 (1:1000; Abcam, Cambridge, UK), anti-caspase-8 antibodies (1:1000; Proteintect, Wuhan, China), anti-BCL2 (1:1000; Proteintect, Wuhan, China), anti-BAX rabbit polyclonal antibody (1:1000; Proteintect, Wuhan, China), anti-cytochrome c (1:1000; Proteintect, Wuhan, China), anti-VDAC1 (1:2000; Proteintect, Wuhan, China), and anti-β-actin (1:2000; Beyotime, Beijing, China). Protein products were analyzed by the Western Bright™ ECL Spray using horseradish peroxidase (HRP)-labelled secondary antibodies (Millipo, USA). The expression levels were normalized to β-actin/VDAC1 levels in corresponding samples that were quantified on a parallel western blot, wherever indicated. Data were analyzed by densitometry using the ImageJ software.
RNA extraction and quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis
The RNA simple Total RNA Kit (TIANGEN) was used to extract total ribonucleic acid (RNA) from the transfected cells. The primers were as follows: β-actin (forward, 5′ ATCATGTTTGAGACCTTCAACA 3′ and reverse, 5′ CATCTCTTGCTCGAAGTCCA 3′) and NOTCH3 (forward, 5′ GCGGGTGGGCAGTGTGTGGAT 3′ and reverse, 5′ CGTCCACGTCGTCCTCACAGTTATCAC 3′). Data were obtained by performing three independent in vitro experiments.
Immunocytochemistry
Stably transduced HEK 293T cells (5×104) were plated in 24-well plates containing poly-L-lysine-coated coverslips. They were fixed using 4% paraformaldehyde for 30 min. After washing thrice with phosphate-buffered saline (PBS), the cells were permeabilized using 0.3% Triton X-100 for 30 min. Nonspecific binding was blocked using 5% bovine serum albumin (BSA) in PBS containing Tween detergent® (BSA-PBST) for 30 min. The cells were incubated with anti-NOTCH3 mouse monoclonal antibodies (H00004854-M02, Novus, USA, 1:200 diluted) overnight at 4°C. After washing twice in 1% BSA-PBST, the cells were incubated with Alexa488-labeled goat anti-mouse secondary antibody (Invitrogen, USA, 1:200 diluted) or Alexa564-labeled goat anti-mouse secondary antibody (Invitrogen, USA,1:200 diluted) for 1 h. Nuclei were dyed with 5μg/ml 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) (Beyotime, Shanghai, China) for 5 min and washed twice with PBS for 5 min to remove any excess dye. Subsequently, the cells on the poly-L-lysine-coated coverslips were mounted using a drop of the Antifade Mounting Medium (Beyotime, Shanghai, China). Subsequently, the cells were imaged on the Olympus Model BX53 fluorescent microscope (Olympus, Tokyo, Japan). For co-staining with Thioflavin S (Thio-S) and NOTCH3 antibody, following fixing with 4% paraformaldehyde and washing with PBS, the cells were stained with Thio-S (1 mg/mL), diluted 1:50 in PBS, for 5 min. The remaining steps are the same as above. Data were obtained by performing three independent in vitro experiments.
Cell counting kit-8 (CCK8) assay
The cells (8×104) were seeded in 96-well plates. After 24 h, 10μL of cell counting kit-8 solution (Dojindo, Japan) was added, and the cells were incubated for 4 h. The absorbance value at 450 nm was detected with a microplate spectrophotometer (Synergy H1, BioTek Instruments, USA). Data were obtained by performing three independent in vitro experiments.
NOTCH3 ECD degradation assay
Stably transduced HEK 293T cells were seeded in 6-well plates. When the cell density reached 80%, the cells were treated with 150μg/mL of cycloheximide (CHX, Sigma-Aldrich, St. Louis, MO, USA), a protein synthesis inhibitor, to follow protein degradation over time. At 0, 3, 6, and 9 h after treatment, the cells were collected and cell lysates were prepared for immunoblotting of NOTCH3ECD using β-actin as a loading control. Data were obtained by performing three independent in vitro experiments.
Mitochondrial transmembrane potential measurement
Mitochondrial membrane potential assay kit with JC-1 (Beyotime, Shanghai, China) was used to analyze mitochondrial membrane potential. Stably transduced HEK 293T cells were seeded in 6-well plates. Next day, after washing once with PBS, 1 mL of cell culture medium and 1 mL of JC-1 working solution were added to the cells. The cells were incubated at 37°C for 30 min. The cells were washed twice with JC-1 (1×), and added 1 mL PBS into each well and blow gently to single cells. Changes in the ΔΨm were analyzed using flow cytometry with flow cytometer (FACS Aria III, BD, USA). The PE and FITC channels were used to detect red JC-1 aggregates in healthy cells and green JC-1 monomers in apoptotic cells. The ΔΨm was represented as the number ratio of the healthy cells with mitochondria that contain JC-1 aggregates to the apoptotic cells with JC-1 monomers. Data were obtained by performing three independent in vitro experiments.
Statistical analysis
SPSS version 22 (IBM, Armonk, NY) was used to conduct statistical analyses. Data were compared by one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test and expressed as the mean±standard deviation (SD). A value was considered statistically significant at p < 0.05. GraphPad Prism 8.0 software was used for statistical analysis and graphing.
RESULTS
Clinical features of two pedigrees
The proband (Fig. 1a, II-5) of F1, a 45-year-old woman with a previous personal history of hypertension and diabetes, was referred to the neurology department because of recurrent ischemic strokes. She experienced her first ischemic stroke at the age of 37 years, which recurred six times. Gradually, she developed gait impairment, aphasia, cognitive impairment, and apathy. Fluid-attenuated inversion recovery (FLAIR) (MRI 3.0T) revealed symmetric confluent WMH, particularly in the temporal pole and periventricular regions, and multiple LI in the periventricular regions (Fig. 1d). Her father developed memory loss at the age of 55 years and died at the age of 60 years (the cause of death was unknown). Sibling II.2 experienced the first ischemic stroke at the age of 40 years with slurred speech and hemiplegia in early stages, subsequent cognitive impairment and gait difficulties. Other family members were in healthy.
The proband (Fig. 1a, II.1) of F2 was referred to the neurology department owing to memory deficits and progressive gait impairment. His first symptom of migraines without aura occurred at the age of 40 years. The proband experienced his first ischemic stroke resulting in dizziness at the age of 66 years, which recurred twice. Gradually, this developed into memory deficits, gait impairment, irritability, and urinary incontinence. Eventually, he finally died from lung disease at the age of 76 years. FLAIR (MRI 3.0T) included WMH of the external capsule and periventricular regions, multiple LI and cerebral microbleeds (Fig. 1d). III.2 was the 45-year-old son of proband F2 who presented with occasional dizziness and enlarged perivascular spaces in basal ganglia on MRI (Fig. 1d).
GOM deposits were observed by analyzing at least six arteriolar vessels in the 2 probands (Fig. 1c). In addition, the atrophy of VSMCs, stenosis of vascular lumen and thickening of vascular walls were observed. The common clinical traits of the two probands are as follows: 1) all conditions developed in middle age, 2) most patients presented with recurrent ischemic strokes and cognitive impairment, 3) FLAIR revealed WMH in the temporal pole or the external capsule, and 4) GOM deposits were observed on skin biopsy.
NOTCH3 mutation analysis and prediction of bioinformatics
We confirmed a de novo heterozygous missense mutation c.1690G > A in NOTCH3 in the two probands, leading to an amino acid exchange (p. Ala564Thr [p. A564T]) (Fig. 1b). Furthermore, through Sanger sequencing, we observed that II.2, III.10, and III.11 of F1 and III.2 and III.3 of F2 had this mutation. However, we did not observe this mutation in the 120 healthy blood donors.
The NOTCH3 mutation (c.1690G > A, p. Ala564Thr) is encoded by exon 11 and located at the 14th of EGFr domain in NOTCH3ECD. The minor allele frequency of this mutation was 0.0001 in the gnomAD and ExAC databases and 0 in Asian population. This mutation has not been reported in the HGMD database. Furthermore, the novel mutation was considered to be pathogenic in 18 of the 22 silico prediction tools by VarCards, and the damaging score was 0.82. The closer the damaging score was to one, the more pathogenic the mutation was. Furthermore, Ala at position 564 in the NOTCH3 protein sequence was highly conservative among different species as analyzed using multiple sequence alignments (Fig. 1e). In addition, the protein structure changed from the Ala to the Thr at position 564 in NOTCH3 protein (Fig. 1f).
The clinical features, load of MRI and GOM deposits of patients with p. A564T mutation were similar to patients with cysteine-affecting NOTCH3 mutation. Therefore, we hypothesized that this mutation may also affect the aggregation and degradation of NOTCH3ECD and ultimately cause cellular dysfunction. Hence, we implemented the following experiments to prove our hypothesis.
The aggregation of NOTCH3 ECD A564T mutation in cells
Currently, the pathogenicity of cysteine-sparing NOTCH3 mutations in CADASIL remains controversial. To evaluate the pathogenic role of p. A564T, we successfully established the stably transduced HEK 293T cell line expressing NOTCH3ECD WT, NOTCH3ECD A564T mutation and a void vector, and we observed the expression of NOTCH3 in NOTCH3ECD A564T and WT group at both the protein and mRNA levels, but not in the void vector control (Fig. 2). It was remarkable that the expression of NOTCH3 in NOTCH3ECD A564T group was higher than that in NOTCH3ECD WT group and void vector control at both the protein (Fig. 2b) and mRNA levels (Fig. 2c). We have observed this phenotype in cysteine-affecting NOTCH3 mutations [18], and an increased expression of NOTCH3 protein was considered to be a compensatory mechanism for mutation-induced loss of function. Furthermore, we observed the allocation of NOTCH3ECD in cells using immunofluorescence. We observed that NOTCH3ECD was not observed in the cytoplasm of void vector control and formed aggregation-like staining in NOTCH3ECD WT group, however, displayed larger aggregated forms in NOTCH3ECD A564T group (Fig. 3a). The immunofluorescent density tended to be significantly higher than NOTCH3ECD WT group and void vector control (Fig. 3b).
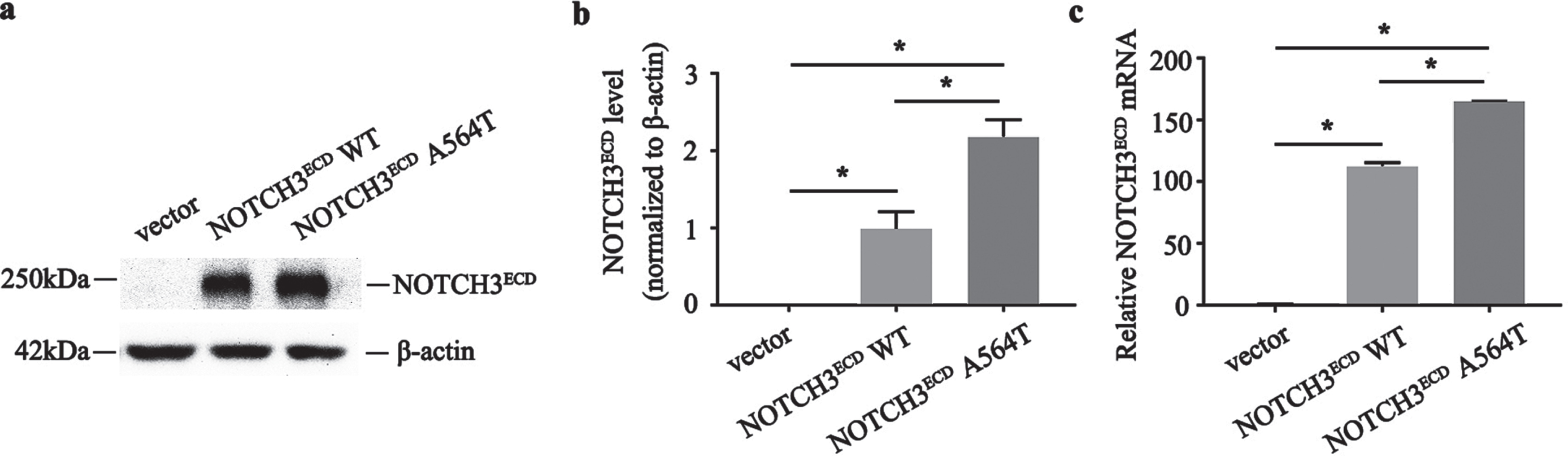
Construction and validation of lentiviral cell lines. a) Protein levels of NOTCH3ECD are detected by western blot, and β-actin is used as a loading control. b) Quantitative analysis of a. c) Quantitative analysis of the NOTCH3ECD messenger ribonucleic acid (mRNA) expression. Data are presented as means±SD. * p < 0.05, n = 3 per group; one - way ANOVA followed by Dunnett’s post hoc test.
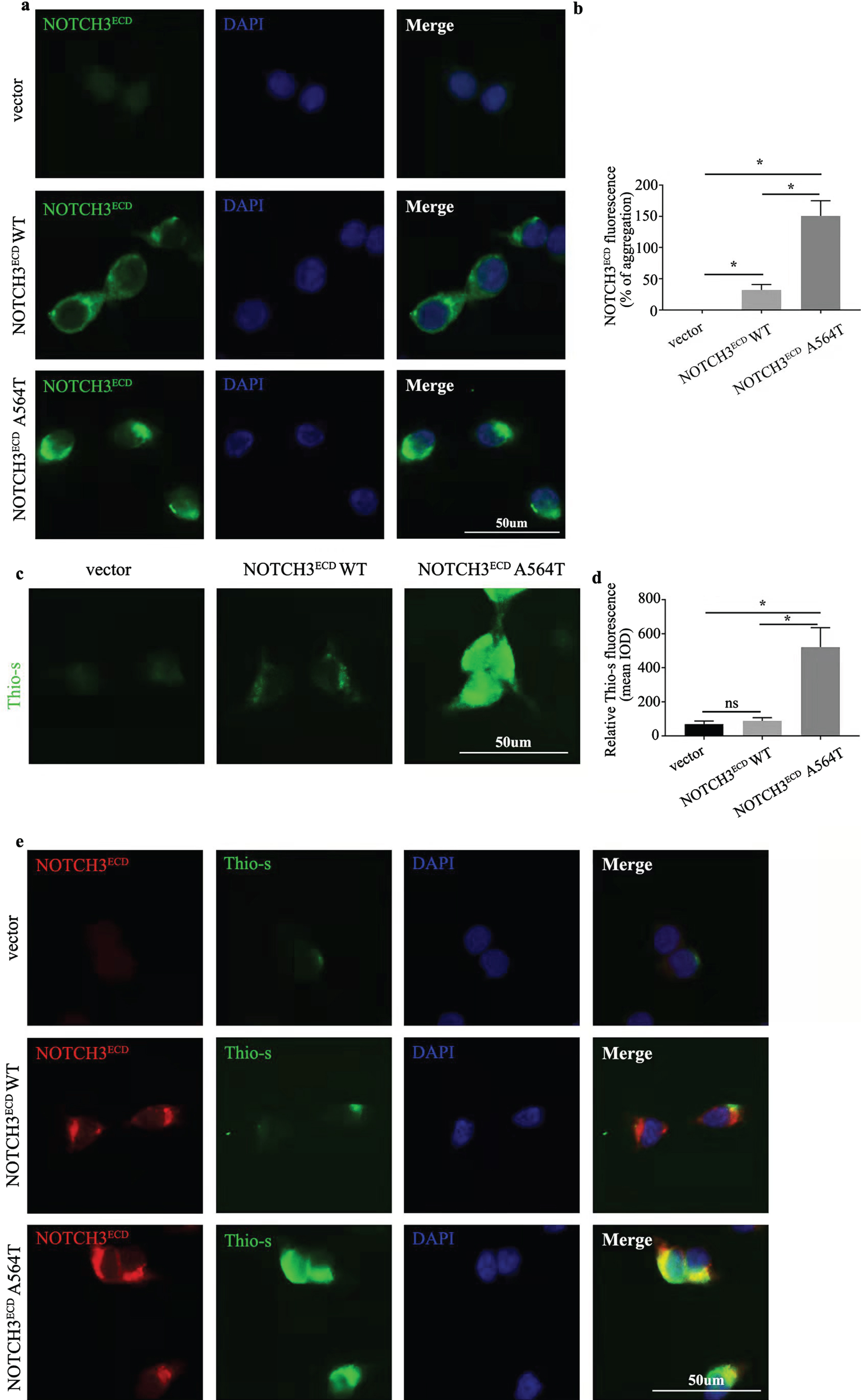
Aggregation and distribution of NOTCH3ECD. a) Immunofluorescence staining exhibits NOTCH3ECD (green) and the nuclei (blue). b) Semi-quantitative analysis of a. Analysis performed using Image Pro Plus software to quantify the mean Integrated Optical Density (IOD) parameter. c) Immunofluorescence staining exhibits Thioflavin-S (Thio-S) positivity (green). d) Semi-quantitative analysis of c. Semi-quantitative analysis of c. Analysis performed using Image Pro Plus software to quantify the mean IOD parameter. e) Double staining exhibits co-localization of aggregated NOTCH3ECD (red) in Thio-S positive (green) aggregates. Data are presented as means±SD. * p < 0.05, n = 3 per group; one - way ANOVA followed by Dunnett’s post hoc test.
We performed Thioflavin-S (Thio-S) staining for evaluating protein aggregation-like in cells. Few Thio-S-positive aggregates were observed in NOTCH3ECD WT group, void vector control and irrelevant transfected 293T cells, while the larger and more Thio-S-positive aggregates formed in NOTCH3ECD A564T group (Fig. 3c, d and Supplementary Figure 2). In order to identify aggre-gates in cells, we performed a co-staining usingThio-S and NOTCH3ECD. The Thio-S-positive agg-regates did not entirely co-located with NOTCH3ECD in NOTCH3ECD WT group; however, the co-localization of thio-s-positive aggregates with NOTCH3ECD was more obvious in NOTCH3ECD A564T group revealing NOTCH3 A564T mutation caused the formation of aggregation-like (Fig. 3e).
Resistance to the degradation of NOTCH3 ECD A564T mutation
The in vitro studies revealed that NOTCH3ECD A564T mutation was more likely to form aggregates as compared with NOTCH3ECD WT. Therefore, in order to investigate the possible reasons for higher aggregation of NOTCH3ECD A564T mutation, CHX pulse-chase experiment was performed. After CHX treatment for 6 h, the NOTCH3ECD WT cells had considerably degraded. However, the cells harboring A564T mutation remained unchanged. The degradation of NOTCH3ECD A564T mutation was lower than that of NOTCH3ECD WT (p < 0.05, Fig. 4). This implied that the NOTCH3ECD with A564T mutation was more difficult to degrade as compared with NOTCH3ECD WT.
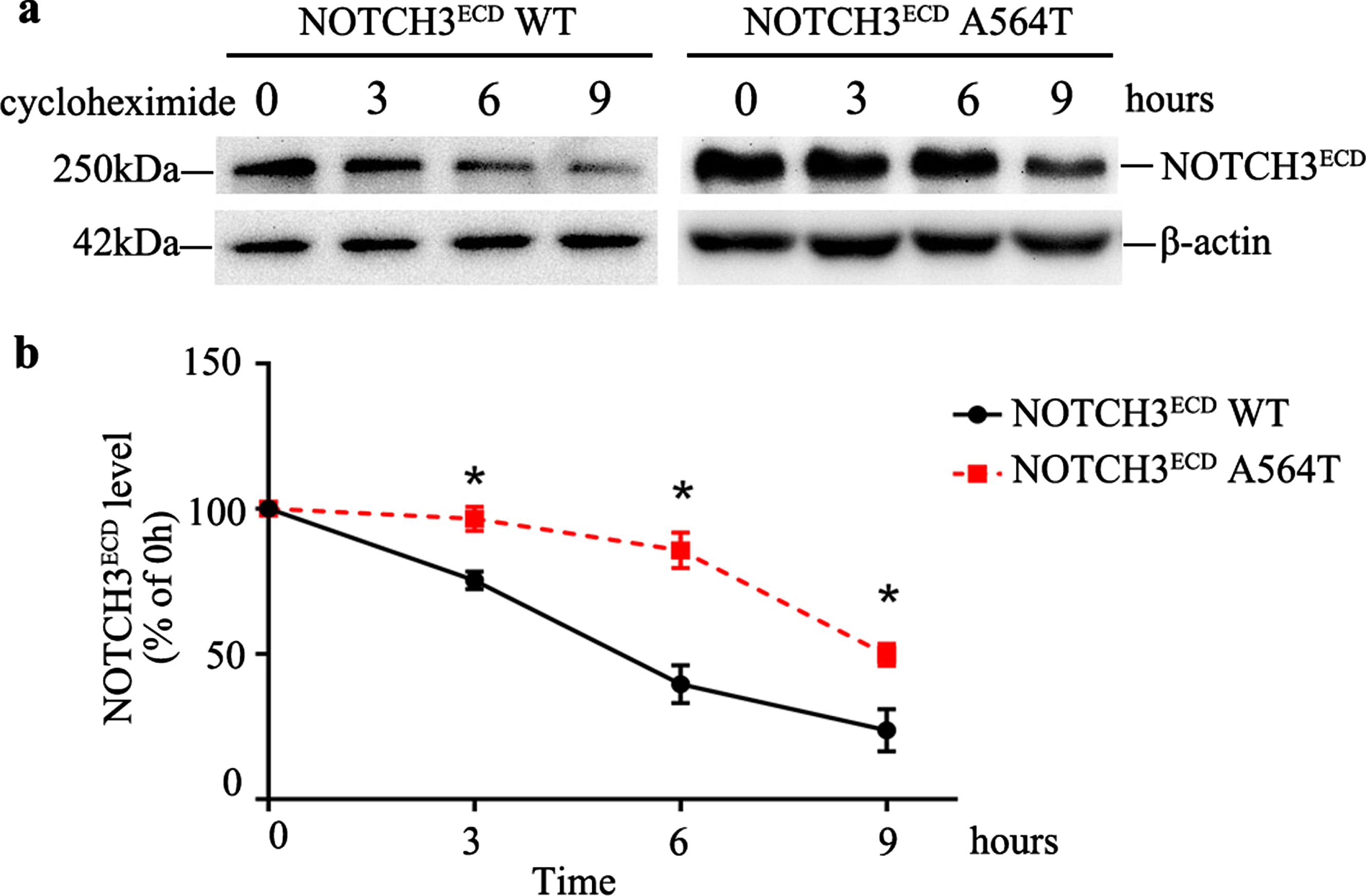
Protein stability analysis and degradation of NOTCH3ECD in cells. a) The protein stability analysis in NOTCH3ECD WT and NOTCH3ECD A564T group. The stably transduced HEK 293T cells are chased with 150μg/mL cycloheximide (CHX) at different time points and detected by western blot, and β-actin is used for loading control. b) Quantitative analysis of a, Data are presented as means±SD. * p < 0.05 versus the NOTCH3ECD WT group, n = 3 per group; one - way ANOVA followed by Dunnett’s post hoc test.
Cell viability decreased with NOTCH3 ECD A564T mutation
We assessed the effects of the aggregation of NOTCH3ECD A564T mutation in HEK 293T cells by CCK8 cell viability assays. As presented in Fig. 5a, the viability in NOTCH3ECD A564T group (77.70±3.43%) significantly decreased as compared with NOTCH3ECD WT group and void vector control (p < 0.001). However, the viability of cells expressing NOTCH3ECD WT was not significantly affected as compared with cells expressing a void vector (p = 0.072).
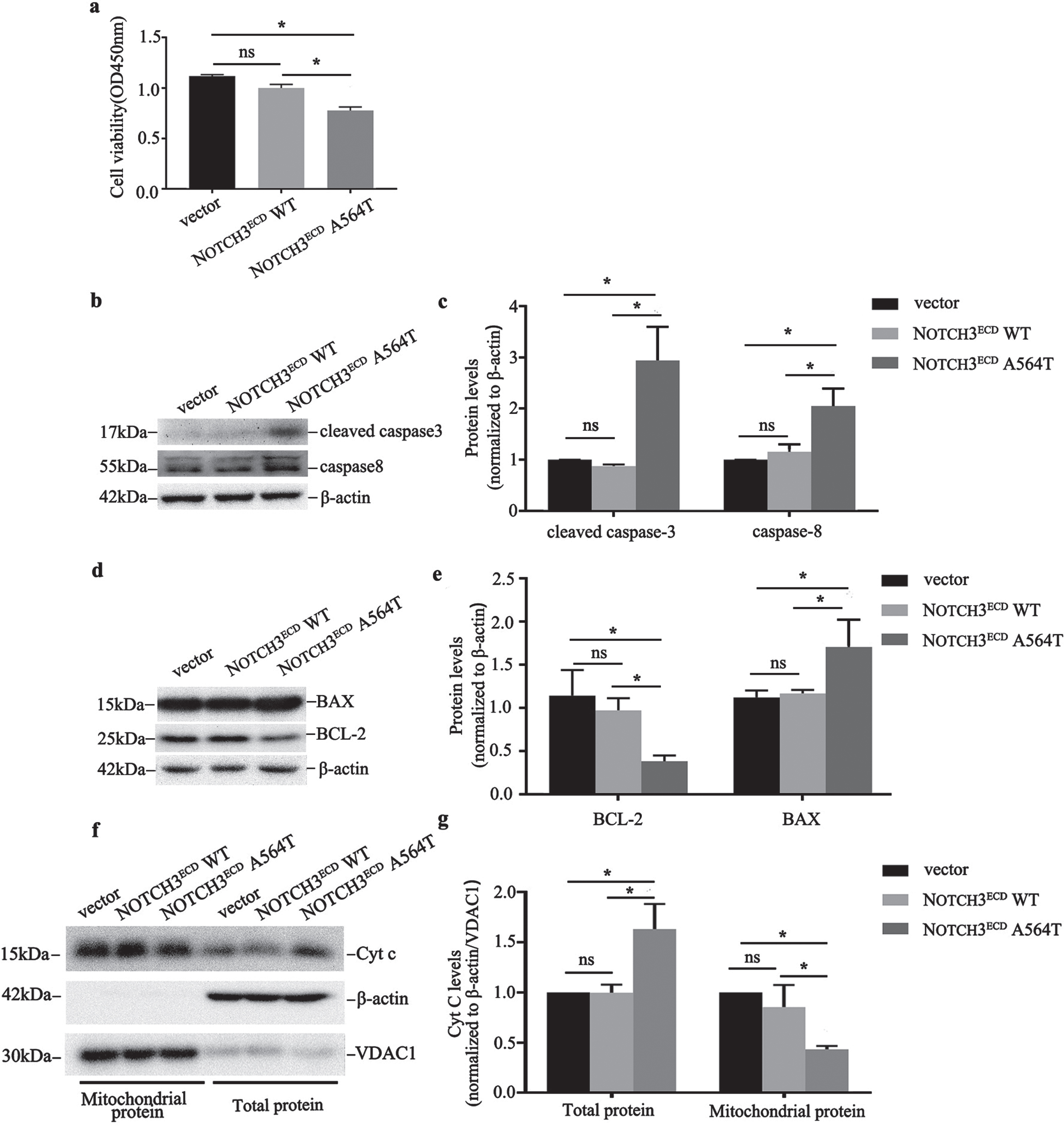
NOTCH3ECD A564T mutation - induced apoptosis. a) Quantitative analysis of cell viability in NOTCH3ECD WT, NOTCH3ECD A564T group and void vector control. b, d, f) Expression of cleaved caspase-3, caspase-8, BCL2, BAX and cytochrome c (Cyt c) are detected by western blot, and β-actin is used for loading control. c, e, g) Quantitative analysis of b, d, f. Data were presented as means±SD, * p < 0.05, n = 3 per group; one - way ANOVA followed by Dunnett’s post hoc test.
Apoptosis increased with NOTCH3 ECD A564T mutation
Furthermore, we assessed the potential mechanism of the NOTCH3ECD A564T mutation, leading to decreased cell viability. Initially, we used western blot to test the apoptosis effector protein caspase-8 and cleaved caspase-3 and we observed that cells overexpressing NOTCH3ECD A564T mutation exhibited significantly higher expression of caspase-8 and cleaved caspase-3 than that exhibited by cells expressing NOTCH3ECD WT and a void vector (Fig. 5b, c). Apoptosis can be initiated by caspase-8, which can activate the mitochondria-mediated caspase3-dependent apoptosis pathway [19]. The BAX and BCL-2 located on the outer mitochondrial membrane are involved in the regulation of cell apoptosis; therefore, we further observed the change in BAX and BCL-2. The results revealed that cells overexpressing NOTCH3ECD A564T mutation exhibited a significantly higher expression of BAX and lower expression of BCL-2 than that exhibited by cells expressing NOTCH3ECD WT and a void vector (Fig. 5d, e).
Mitochondrial impairment in cells with NOTCH3 ECD A564T mutation
Previous studies have demonstrated that upregulation of BAX and downregulation of BCL-2 may affect the changes of ΔΨm and mitochondrial permeability transition pore [20]. We further investigated mitochondrial function by detecting ΔΨm in the stably transduced HEK 293T cells. As indicated in Fig. 6a, the P2 gate represents the healthy cells with mitochondria that contain JC-1 aggregates and the P3 gate represents the apoptotic cells with JC-1 monomers. The number ratio of the healthy cells with mitochondria that contain JC-1 aggregates to the apoptotic cells with JC-1 monomers, which represents the change of ΔΨm, in NOTCH3ECD A564T group was obviously decreased as compared with NOTCH3ECD WT group and void vector control (p < 0.05) (Fig. 6b). In addition, cytochrome c in cells expressing NOTCH3ECD A564T mutation was significantly higher than that present in cells expressing NOTCH3ECD WT and a void vector (p < 0.05); however, cytochrome c in mitochondria was significantly lower (p < 0.05) (Fig. 5f, g). This indicated that the mitochondrial permeability transition pore may be open and cytochrome c in the mitochondria was transported to the cytoplasm, thus activating the caspases-3-dependent apoptotic pathway.
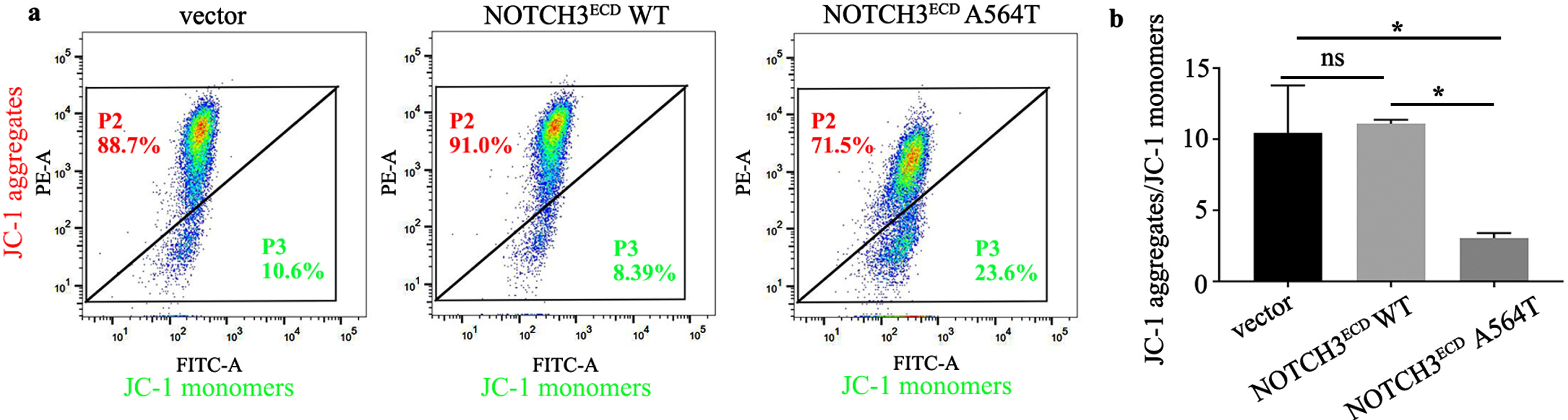
Analysis of the mitochondrial membrane potential (ΔΨm) in NOTCH3ECD WT, NOTCH3ECD A564T group and void vector control by JC-1 staining. a) The P2 gate represents the healthy cells with mitochondria that contain JC-1 aggregates and the P3 gate represents the apoptotic cells with JC-1 monomers. b) The ΔΨm of was represented as the ratio of the number of the healthy cells with mitochondria that contain JC-1 aggregates to the apoptotic cells with JC-1 monomers. * p < 0.05; one - way ANOVA followed by Dunnett’s post hoc test.
DISCUSSION
In our study, a de novo mutation (c.1690G > A, p. A564T) in exon 11 of NOTCH3 in two pedigrees was reported for the first time. Based on in vitro experiments, we observed that this mutation was likely pathogenic. First, we determined that two probands with symptoms of CADASIL harbored the cysteine-sparing NOTCH3 mutation. Second, GOM deposits were discovered on skin biopsy in the two probands. Third, the results of the in vitro experiments revealed that the A564T mutation caused abnormal aggregation and resistance to the degradation of NOTCH3ECD. Lastly, we observed that A564T mutation may influence cell viability and mediate cell apoptosis by mitochondrial dysfunction. In this study, we may attain a new breakthrough point to explain the pathogenesis of a novel cysteine-sparing NOTCH3 mutation (p. A564T) in CADASIL.
Currently, the pathogenicity of cysteine-sparing NOTCH3 mutations remains controversial. Muiño [21] suggested that cysteine-sparing NOTCH3 mutations that meet the following criteria can be potentially pathogenic: 1) there are the classic symptoms of CADASIL, 2) there is diffuse WMH on FLAIR, 3) other exon pathogenic mutations of NOTCH3 have been excluded, 4) mutations are not polymorphic, and 5) skin biopsy reveals GOM deposition. The A564T mutation may be a pathogenic mutation instead of a polymorphism. Initially, two probands presented an archetypical clinical phenotype [3] and the specific MRI findings [22], despite the presence of vascular risk factors. Although the previous studies have demonstrated that patients with CADASIL have no vascular risk factors [23], the absence of vascular risk factors was not mentioned in the current diagnosis of CADASIL in Japan [24]. Furthermore, a prospective cohort study [25] suggested that the cardiovascular risk factors may modulate phenotype. Smoking was associated with an earlier onset of stroke and the onset of dementia in addition to clinical progression in NOTCH3 mutation carriers. A high risk of cardiovascular disease is related to cognitive impairment and lesion load on MRI [26]. Based on the classification of genetic mutations of the American College of Medical Genetics and Genomics (ACMG) [27], the A564T mutation is considered to be likely pathogenic (PM1, PM2, PP1, and PP3). Co-segregation with disease further supported its clinical significance that the A564T mutation was absent in the healthy older siblings and control genomes; however, the ailing brother and son were carriers.
In our study, GOM deposits were observed in the skin biopsies. GOM deposits are a major pathological feature of CADASIL. In CADASIL patients, GOM deposits comprise NOTCH3ECD and extracellular matrix proteins. In our study, we observed that the NOTCH3 A564T mutation resulted in the increased NOTCH3 expression and the formation of aggregative proteins, this phenotype had observed previously [18]. These A564T mutant aggregates were mainly localized in the cells, but not the extracellular region. Duering et al. [7] found spontaneous extracellular aggregation using a truncated 25kDa protein fragment of NOTCH3-EGF1–5; however, neither the NOTCH3ECD nor the shorter fragment of NOTCH3-EGF1–15 was observed in the extracellular region. They suggested that upon overexpression of larger NOTCH3 structures, the complex folding processes of NOTCH3ECD proteins enriched in EGFr may result in a great burden for the folding machinery in the cells, ultimately resulting aggregation of misfolded proteins in the cells. However, GOM formation was observed out of the VSMCs in CADASIL patients and transgenic mice, which needs further research. In addition, the aggregation of NOTCH3ECD in transfected cells also indicated that this mutant was prone to aggregation. Wollenweber et al. [16] using single-particle assay show that cysteine-sparing mutations, such as D80G, exhibit multimerization tendencies similar to those due to cysteine mutations. They speculated two possible mechanisms underlying the multimerization behavior of cysteine-sparing mutations. In one case, the exchange of cysteine-sparing amino acids could lead to the destruction or recombination of disulfide bonds, thereby resulting in the formation of unpaired cysteine residues. These could therefore induce conformational changes, which in turn promoted polymerization through disulfide bonds. Our experimental findings further suggested that the accumulation of NOTCH3ECD A564T could be due to its resistance to degradation. This opens a new avenue for disease modification therapy in CADASIL. Regarding the aggregation of NOTCH3 mutants, some scholars suggest that aggregation and accumulation of mutant NOTCH3 are associated with dysfunction in autophagy-lysosomal pathways in CADASIL VSMCs [18]. However, a clear conclusion on the mechanism of mutant NOTCH3 aggregation is lacking. Perhaps, both anti-degradation of mutant NOTCH3 aggregation and dysfunction of the NOTCH3 degradation induced by mutations are involved in the pathogenesis ofCADASIL.
NOTCH3, a cell-surface receptor involved in the determination of cell fates, influences cell proliferation and apoptosis [28]. Recent studies show that NOTCH3 has a protective effect in VSMCs apoptosis, as it inhibits FasL-induced apoptosis by upregulating C-FLIP in an ERK/MAPK dependent pathway [29]. Formichi et al. [30] report that peripheral blood lymphocytes (PBLs) and fibroblasts from CADASIL patients exhibit more significant responses to 2-deoxyd-ribo-induced apoptosis than the normal subjects, suggesting that the NOTCH3 mutation is an important trigger for apoptosis. PBLs from CADASIL patients show high levels of apoptosis even without apoptotic stimulation. Hence, the cells of CADASIL patients are physiologically predisposed to apoptotic cell death and NOTCH3 may be involved in the regulation of endogenous apoptotic pathways. This was consistent with our findings as cell viability was significantly decreased and cells apoptosis was enhanced in A564T NOTCH3 mutated cell. In addition, Formichi report that PBLs in CADASIL patients show a higher degree of mitochondrial membrane depolarization than the controls. Mitochondria are key cell organelles, which play a vital role in energy generation and regulation of cell metabolism. The structural integrity and normal functioning of mitochondria are necessary for cell survival and apoptosis. Mitochondrial dysfunction may result from oxidative damage caused by free radicals or direct damage from the accumulation toxic metabolites in certain metabolic diseases. Alterations in proteins having non-natural conformations and aggregation are often associated with the endoplasmic reticulum and stress, which lead to mitochondrial dysfunction and excessive production of ROS [31]. Some studies show that oxidative stress affects cell metabolism by damaging mitochondrial function [32]. At the molecular level, vascular abnormalities in CADASIL are associated with NOX5-induced ROS, endoplasmic reticulum stress response, and activation of Rho kinase [8]. NOX5 localizes on the outer mitochondrial membrane and is an important NADPH oxidase. In CADASIL, whether oxidative stress is related to mitochondrial dysfunction, remains unclear. The results of our study showed that ΔΨm of HEK 293T cells with A564T mutation was attenuated and the degree of mitochondrial membrane depolarization was higher as compared to the NOTCH3ECD WT cells, which indicated a higher aberrant mitochondria, consistent with the findings of Viitanen [12]. However, VSMCs in CADASIL in their study showed reduced proliferation without a significant decrease in cell viability and intracellular ROS content at the basal level. Currently, studies on the relationship between mitochondrial dysfunction and VSMCs have only investigated a few mutations in CADASIL; however, more than 230 mutations in CADASIL are known. Thus, the mechanism underlying mitochondrial dysfunction in CADASIL requires further study.
Both BAX and BCL-2 belong to the BCL-2 family of proteins and regulate cell survival and apoptosis through mitochondrial pathway. BAX promotes apoptosis, while BCL-2 prevents apoptosis. The ratio of BAX/BCL-2 is an essential factor in determining the inhibitory effect of cell apoptosis. Mitochondria is the main organelle that controls cell apoptosis. In neurological diseases, the initiation of mitochondria-mediated apoptosis can be caused by both internal and external cascades. Internal cascades are triggered primarily by cellular stress or damage. During this process, the BCL-2 family of proteins regulates increased mitochondrial membrane permeability, which activates cell apoptosis and allows for the release of cytochrome c factors into the cytoplasm. All these proteins are involved in the mitochondrial pathology underlying neurological disorders [33]. Therefore, we hypothesized that NOTCH3 mutations may induce apoptosis through the mitochondrial-mediated pathway. We observed that the HEK 293T cells carrying the A564T mutation expressed high BAX levels, low BCL-2 levels, and increased BAX/BCL-2 ratio as compared to the WT cells in our study. More importantly, cytochrome c levels were elevated in the cytoplasm, and attenuated in the mitochondria in our study. The release of cytochrome c from mitochondria into the cytoplasm is a key step in the process of apoptosis. In addition, NOTCH3 signaling can inhibit apoptosis by upregulating C-FLIP to inhibit the connection with caspase-8 and Fas receptor complex [29]. Therefore, we evaluated the effector caspases. In our study, cleaved caspase-3 and caspase-8 levels were higher in HEK 293T cells carrying the A564T mutation. Moreover, the loss of VSMCs in the CADASIL mouse brain was very significant, and the activity of caspase-3 in VSMCs was obviously increased. After the treatment of stem cell factor joint granulocyte colony-stimulating factor (SCF+G-CSF) in CADASIL mice, GOM deposits in blood vessels of the brain cortex was inhibited, along with the loss of VSMCs in brain arteriolar walls and the activity of caspase-3 had a significant reduction [34]. These suggests that GOM deposits may cause denaturation and loss of VSMCs in CADASIL. Taken together, the results suggest that increased apoptosis in CADASIL may be related to mitochondrial dysfunction, however, the specific molecular mechanism needs to be investigated in the future. Based on these results, we also proposed a possible schematic model of A564T-mutation-induced apoptosis through the mitochondrial-mediated pathway (Fig. 7).
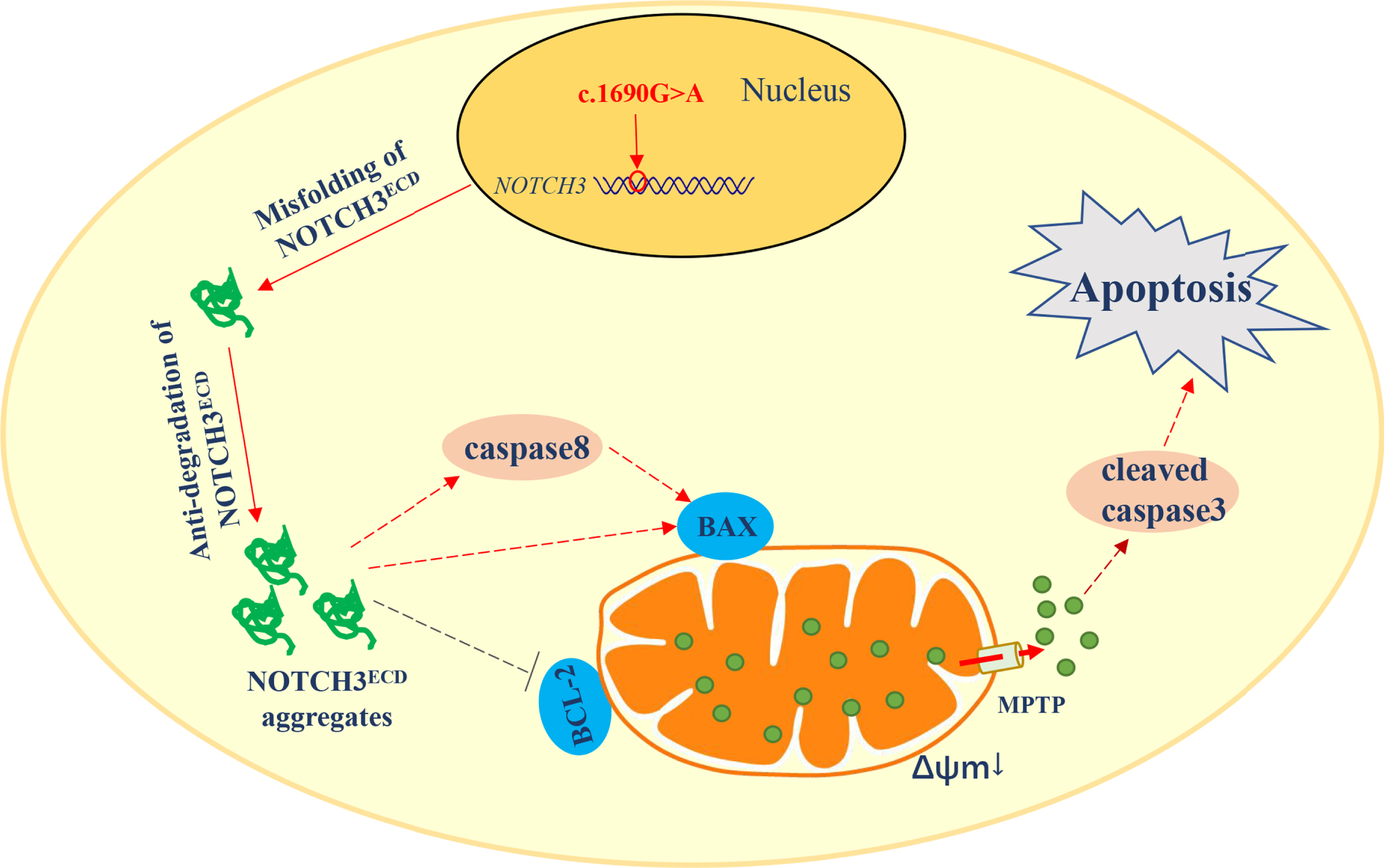
Schematic of the hypothesis that A564T-mutation-induced apoptosis through the mitochondria - mediated pathway in stably transduced HEK 293T cells. NOTCH3 (c.1690G > A) mutation leads to misfolding and anti-degradation of NOTCH3ECD in cells. NOTCH3ECD aggregates may activate caspase-8 and BAX, while inhibit BCL-2 located on mitochondrial transmembrane. Moreover, activated caspase-8 may further promotes role of BAX in mitochondria, affecting the ΔΨm and the mitochondrial permeability transition pore (MPTP) of mitochondria. Cytochrome c (Cyt c) liberates from the mitochondria into cytoplasm through the damaged mitochondrial membrane and may activate the effector caspase-3 to release cleaved caspase-3, inducing apoptosis.
However, there are some limitations to this study. The in vitro assays were based on the HEK 293T cell lines rather than patient-derived VSMCs or fibroblasts. Patient-derived VSMCs or fibroblasts were not available due to the immediate glutaraldehyde fixation and upon embedding followed in routine clinical operations. However, we were able to observe the possible presence of mitochondrial dysfunction in HEK 293T cells carrying the A564T mutation. In addition, complementary experiments are needed to investigate the mechanisms underlying mitochondrial dysfunction in CADASIL.
In conclusion, our study described a de novo cysteine-sparing NOTCH3 mutation (c.1690G > A) for the first time, which broadened the mutation spectrums of CADASIL. The aggregation of NOTCH3ECD A564T mutation may be associated with more difficult degradation of the mutant, and the aggregation may produce toxic effects to induce apoptosis through mitochondrial-mediated pathway. Therefore, we speculated that mitochondrial dysfunction may hopefully explain the pathogenesis of cysteine-sparing NOTCH3 mutation in CADASIL. Our study may provide a new strategy for clinical therapy of CADASIL in the future. However, the molecular mechanism of mitochondrial dysfunction in CADASIL needs to be further studied.
Footnotes
ACKNOWLEDGMENTS
We would like to thank all subjects for their participation, as well as all further contributors for their work and support for this study.
This research was supported by the National Natural Science Foundation of China (Grants 81873727) and Key Science and Technology Program of Henan Province, China (201701020, 20210231008).