
Abstract
Background:
Cerebral amyloid angiopathy is a cerebrovascular disease directly implicated in Alzheimer’s disease pathogenesis through amyloid-β deposition. Growing evidence has shown a pivotal role of chronic neuroinflammation both in cerebral amyloid angiopathy and Alzheimer’s disease.
Objective:
The aim of this study was to investigate whether circulating levels of the complement 3, a crucial component of the innate immune system, are increased in patients with cerebral amyloid angiopathy.
Methods:
Serum complement 3 levels were retrospectively measured by a sandwich enzyme-linked immunosorbent assay in a single-center cohort of patients with mild cognitive impairment. The diagnosis of cerebral amyloid angiopathy was based on the modified Boston criteria. Logistic regression analysis was performed to identify the predictive factors for cerebral amyloid angiopathy.
Results:
We analyzed 55 mild cognitive impairment patients (mean age [standard deviation]: 76.3 [6.8] years; 33 [60% ] men). Complement 3 levels were significantly increased in cerebral amyloid angiopathy patients (n = 16) compared with those without cerebral amyloid angiopathy (n = 39) (median [interquartile range]: 0.43 [0.34–0.65] versus 0.35 [0.25–0.45], respectively; p = 0.040). Univariate and multivariate logistic regression analysis revealed that increased complement 3 levels were significantly associated with cerebral amyloid angiopathy. After selection of the best predictive model using stepwise selection, complement 3 was preserved as a significant independent predictive factor for cerebral amyloid angiopathy (odds ratio per 0.1 unit/mL increase [95% confidence interval]: 1.407 [1.042–1.899]; p = 0.026).
Conclusion:
Complement activation may play a pivotal role in cerebral amyloid angiopathy. Complement 3 may be a novel diagnostic biomarker for cerebral amyloid angiopathy.
INTRODUCTION
Cerebral amyloid angiopathy (CAA) induces various forms of cerebral infarcts and hemorrhages associated with accumulation of amyloid-β (Aβ) in the walls of cerebral arteries; there is resulting acceleration of cognitive impairment in Alzheimer’s disease (AD) [1]. Cerebrovascular Aβ accumulation is derived from neurons [2] and is closely associated with impaired Aβ clearance from the brain via the intramural periarterial drainage route [2]. This drainage impairment contributes to CAA, whereas vascular Aβ accumulation can accelerate vascular injury leading to further Aβ elimination failure from the brain [2, 3]. The damaged vascular endothelium can produce proinflammatory cytokines that magnify neuroinflammation, glial activation, and secondary injury, leading to blood–brain barrier breakdown, white matter injuries, and cortical hemorrhages [4, 5]. Therefore, early intervention strategies against CAA would be effective for preventing progression of cognitive impairment in patients with AD [2, 3].
Neuroinflammation is recognized as a key component in CAA and has been mainly demonstrated by the discovery of microglial Aβ binding and complement activation in human autopsy material, as well as from animal and in vitro studies [6–10]. Microglia are the resident immune and phagocytic cells of the nervous system. A transgenic rat model of CAA (rTg-DI) exhibited increased numbers of activated microglia, which was accompanied by expression of complement cascade components [9]. In particular, a robust increase in expression of complement 3 (C3) was observed from the early phase of CAA [9]. C3 expression was also increased in Tg-SwDI mice, another animal model of CAA [8]. In human brains, complement fragment C3d was colocalized with Aβ in the cerebral arteries and capillaries, with their expression levels positively correlated with the amount of Aβ present [6, 7]. Notably, single-cell RNA sequencing of nine AD brains, of which five had CAA, demonstrated much higher expression of C3 in microglia than any other component of the complement cascade [11]. Furthermore, in control brains, C3 expression was highest in microglia, with approximately five-fold higher levels than astrocytes and negligible expression in endothelial and mural cells. Intriguingly, in AD brains, microglial C3 expression was elevated three-fold but decreased in astrocytes compared with control brains, suggesting a close association of microglial C3 with AD and CAA pathogenesis [11]. However, it remains unclear whether the complement cascade is activated in living CAA patients, especially in the early phase. Herein, we examined C3 levels in the blood of CAA patients with mild cognitive impairment (MCI).
METHODS
Study design
This single-center cross-sectional study was performed at the National Cerebral and Cardiovascular Center (NCVC) and conducted in accordance with Declaration of Helsinki standards and after approval by the local ethics committee (M29-166-5). We included MCI patients who fulfilled the following criteria: 1) signed a comprehensive NCVC biobank consent form and 2) who received a Clinical Dementia Rating (CDR) from October 2017 to December 2018. The diagnosis of MCI was based on the global CDR score of 0.5 and the core clinical criteria stated in the National Institute on Aging-Alzheimer’s Association classification for MCI [12]. All patients received blood tests and MRI evaluation to exclude the potential for dementia caused by a treatable condition such as a brain tumor. Pure vascular MCI patients were not included in this study because we did not perform a CDR in patients with cognitive impairment caused solely by cerebrovascular disease.
Clinical assessment
Clinical data were collected from patients’ medical records. The CDR was based on a semi-structured interview with the patient and a caregiver who could provide an index of global functioning [13]. Trained psychologists blinded to the clinical information scored the patients in each of six cognitive domains (memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care). We measured the global CDR score and the CDR-sum of boxes (CDR-SB). The global CDR was scored using the rule reported by Morris [13], while the CDR-SB was the total of each domain score [14].
The concentration of the native form of C3 was measured by a sandwich enzyme-linked immunosorbent assay [15, 16]. We used frozen serum, which was collected within 6 months before or after the CDR. The median (interquartile range) interval was 2 (1–30) days. The assay was independently performed at Molecular and Clinical Bioinformatics Inc. (Tsukuba, Japan).
Genotyping of the APOE gene was performed using a fully automated gene analysis system (LightCycler 96; Roche, Basel, Switzerland), as previously reported [17]. Briefly, the APOE ɛ2 allele was recognized by rs429358-T and rs7412-T, the APOE ɛ3 by rs429358-T and rs7412-C, and the APOE ɛ4 by rs429358-C and rs7412-C. The primer sequences for rs429358 and rs7412 were 5’-CAAGGAGCTGCAGGCGG-3’ (forward) and 5’-CAGCTCCTCGGTGCTCTG-3’ (reverse), and 5’-CGCAAGCTGCGTAAGCG-3’ (forward) and 5’-CGCGGATGGCGCTGAG-3’ (reverse), respectively. The probe sets were 5’-GGACGTGTGCGGCCG-3’ for rs429358-T, 5’-GGACGTGCGCGGCCG-3’ for rs429358-C, 5’-CTGCAGAAGCGCCTGGC-3’ for rs7412-C, and 5’-CTGCAGAAGTGCCTGGC-3’ for rs7412-T. We used patients’ DNA stored at the NCVC biobank.
MRI evaluation was performed by trained neurologists blinded to the clinical information. Lacunar infarcts were defined as rounded or ovoid hypointense lesions of 3–20 mm in diameter, with a hyperintense rim, which occurred in the basal ganglia, thalamus, internal and external capsule, centrum semiovale, and brainstem. Periventricular hyperintensities and deep white matter hyperintensities were scored using the Fazekas scale [18]. Lacunar infarcts and white matter hyperintensities were evaluated using fluid-attenuated inversion recovery images. Cerebral microbleeds (CMBs) were defined as rounded, hypointense foci up to 10 mm in diameter on T2*-weighted images [19]. Either 1.5 T (Magnetom Sonata; Siemens Medical Solutions, Erlangen, Germany) or 3.0 T (Magnetom Verio or Spectra; Siemens Medical Solutions) scanners were used. Probable or possible CAA were diagnosed according to the modified Boston criteria [20]. Briefly, probable CAA requires evidence of multiple strictly lobar hemorrhages, or single lobar hemorrhages and superficial siderosis. Possible CAA requires single lobar hemorrhages or superficial siderosis.
Statistical analyses
Unless otherwise noted, the data are presented as the mean and standard deviation or the median (interquartile range) for continuous variables, and as numbers and percentages for categorical variables. The Student’s t test or Mann–Whitney U test was used to analyze continuous data, while the χ2 or Fisher exact test was used for categorical data. Univariate and multivariate logistic regression models were applied to calculate the odds ratio and 95% confidence interval for identifying predictive factors for probable or possible CAA. Stepwise selection with a p-value of 0.1 for backward elimination was used to select the most significant predictors. Strictly lobar CMBs and superficial siderosis were excluded in the adjustment factors because they are described in the modified Boston criteria for CAA diagnosis. Two-sided p-values <0.05 were considered statistically significant. All statistical analyses were performed with statistical software (SPSS version 26; IBM Corp., Armonk, NY, USA).
RESULTS
Between October 2017 and December 2018, 87 patients received CDR, 59 of whom were diagnosed with MCI according to the National Institute on Aging-Alzheimer’s Association core clinical criteria and a global CDR score of 0.5 (Fig. 1). Of these patients, 55 provided informed consent for participation in the NCVC biobank project.
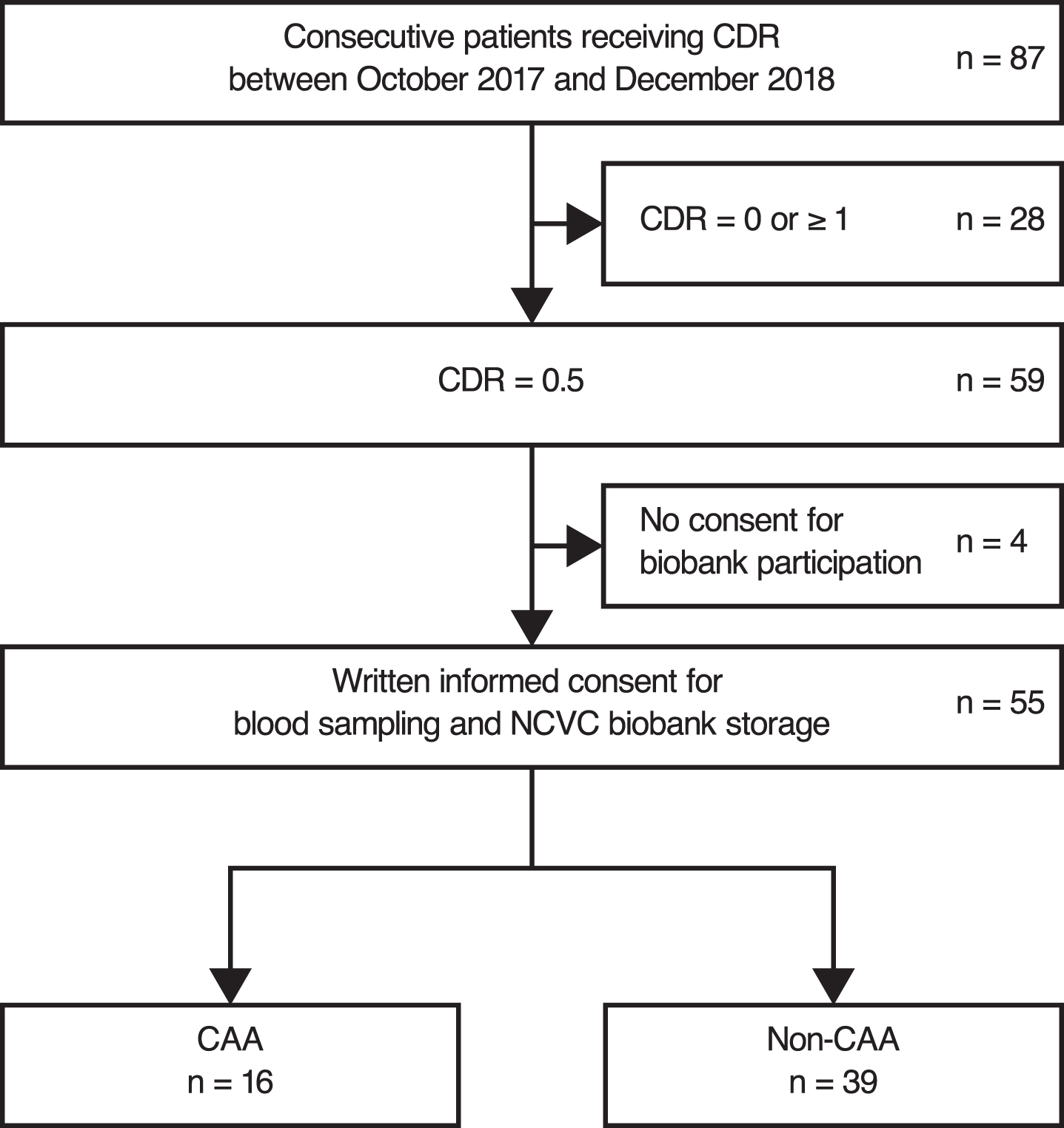
Patient selection. CAA, cerebral amyloid angiopathy; CDR, Clinical Dementia Rating; NCVC, National Cerebral and Cardiovascular Center.
The clinical profiles of the patients are described in Table 1. The mean (standard deviation) age was 76.3 (6.8) years, with 33 (60%) men. CMBs and superficial siderosis were observed in 29 (53%) and 13 (24%) patients, respectively. Based on the modified Boston criteria, four (7%) and 12 (22%) patients presented with possible and probable CAA, respectively. The number of CMBs was significantly higher in CAA patients compared with patients without CAA (3.5 [1.0–37.5] versus 0.0 [0.0–1.0], respectively; p < 0.001). Superficial siderosis was more common in CAA patients compared with patients without CAA (nine [56% ] versus four [10% ] patients, respectively; p < 0.001), while lacunar infarcts were less frequent in the CAA group (three [19% ] versus 19 [49% ] patients, respectively; p = 0.039). Of the 22 patients with lacunar infarcts, CMBs in deep brain regions were observed in 11 (50%) patients. Chronic changes of intracerebral hemorrhage were shown in five patients (31%) with CAA and five patients (13%) without CAA (p = 0.134).
Clinical characteristics
Data represent mean±standard deviation, median (interquartile range), or number (percentage). C3, complement 3; CAA, cerebral amyloid angiopathy; CDR-SB, Clinical Dementia Rating-sum of boxes; CMBs, cerebral microbleeds; DWMH, deep white matter hyperintensities; PVH, periventricular hyperintensities.
The median C3 level in all MCI patients was 0.38 (0.29–0.50) units/mL. The C3 level was significantly increased in CAA patients compared with patients without CAA (0.43 [0.34–0.65] versus 0.35 [0.25–0.45], respectively; p = 0.040). The C3 levels were equivalent between the possible and probable CAA patients (Fig. 2).
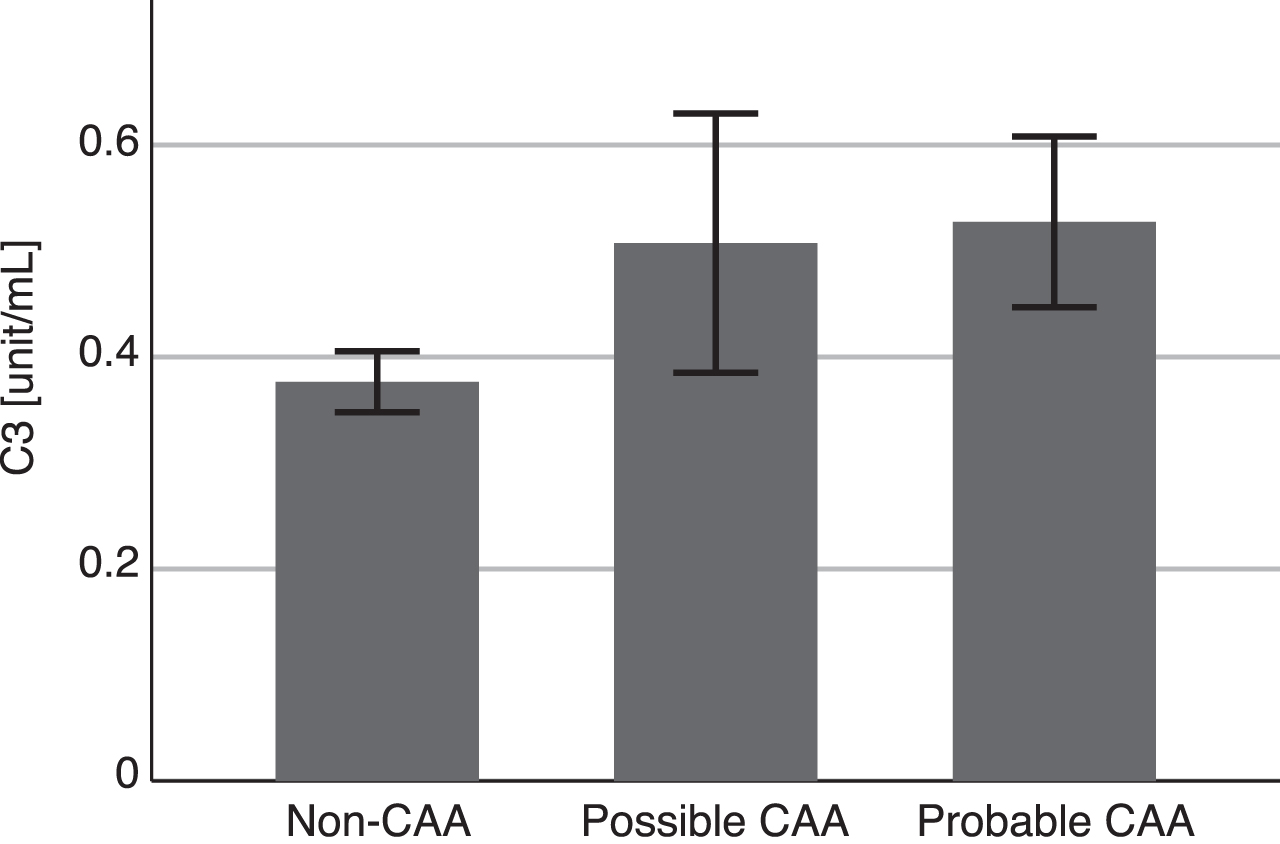
Blood complement 3 levels. Data are mean±standard error of the mean. The median (interquartile range) complement 3 (C3) level was 0.35 (0.25–0.45) in patients without cerebral amyloid angiopathy (non-CAA, n = 39), 0.47 (0.30–0.76) in patients with possible CAA (n = 4), and 0.42 (0.34–0.64) in patients with probable CAA (n = 12). There were no significant differences in C3 levels between the groups (p = 0.120).
Univariate logistic regression analyses showed that CAA was significantly predicted by a higher blood C3 level (odds ratio per 0.1 unit/mL increase [95% confidence interval], 1.365 [1.024–1.820]; p = 0.034) and absence of lacunar infarcts (0.243 [0.060–0.989]; p = 0.048) (Table 2). Statistical significance persisted after adjusting for age, sex, APOE ɛ4, scores on CDR-SB, and number of CMBs (C3:1.435 [1.021–2.017]; p = 0.038; lacunar infarcts: 0.106 [0.016–0.714]; p = 0.021). After selection of the best predictive model using stepwise selection, both C3 and lacunar infarcts were preserved as significant independent predictive factors for CAA (C3:1.407 [1.042–1.899]; p = 0.026; lacunar infarcts: 0.203 [0.044–0.925]; p = 0.039). Receiver operating characteristic analysis of blood C3 level for the prediction of CAA showed an area under the curve (95% confidence interval) of 0.68 (0.53–0.83).
Predictors of cerebral amyloid angiopathy based on univariate and multivariate logistic regression analysis
C3, complement 3; CDR-SB, Clinical Dementia Rating-sum of boxes; CMBs, cerebral microbleeds; OR, odds ratio; 95% CI, 95% confidence interval.
DISCUSSION
In this retrospective study of MCI patients, we found that higher levels of C3 in the blood independently predicted the presence of CAA. An absence of lacunar infarcts was also associated with CAA, which is likely attributable to the comorbidity of lacunar infarcts and deep CMBs. In the current study, 50% of patients with lacunar infarcts presented with deep CMBs. Lacunar infarcts and deep CMBs are well established as imaging markers for hypertensive arteriopathy, but not for CAA [3].
Increased C3 levels are closely associated with chronic inflammation [21–23]. The complement system is a phylogenetically ancient cascade system that has a major role in innate and adaptive immunity [24]. The pivotal complement factor C3 is the convergence point for the classical, lectin, and alternative pathways of complement activation [24]. The function of C3 is regulated by conformational changes induced by sequential proteolytic cleavages [24]. Cleavage of native C3 generates C3a and C3b. Native C3 itself is biologically inactive. However, C3b can bind covalently to the target surface including Aβ via the thioester [10, 24]. Activated microglia express the complement receptor CR3 and can thus bind the C3b-Aβ complex [6, 7]. Activated microglia relocate Aβ to the capillary walls, which may be associated with increased C3 concentration in CAA patients (Fig. 3A) [6, 7]. The elevated C3 levels may also relate to engulfment of cerebrovascular Aβ by microglia around the capillary, which initiates a cascade of neuroinflammation (Fig. 3B). C3a binds to the C3a receptor located in the basolateral surface of brain endothelial cells, inducing vascular inflammation and impairing blood–brain barrier function [25]. C3 is also expressed by perivascular macrophages [11]. Some Aβ can be taken up by perivascular macrophages [26], which may induce neuroinflammation within the intramural periarterial drainage pathways, leading to increased C3 levels in the blood (Fig. 3C). Further studies are warranted to investigate the role of the complement cascade in CAA.
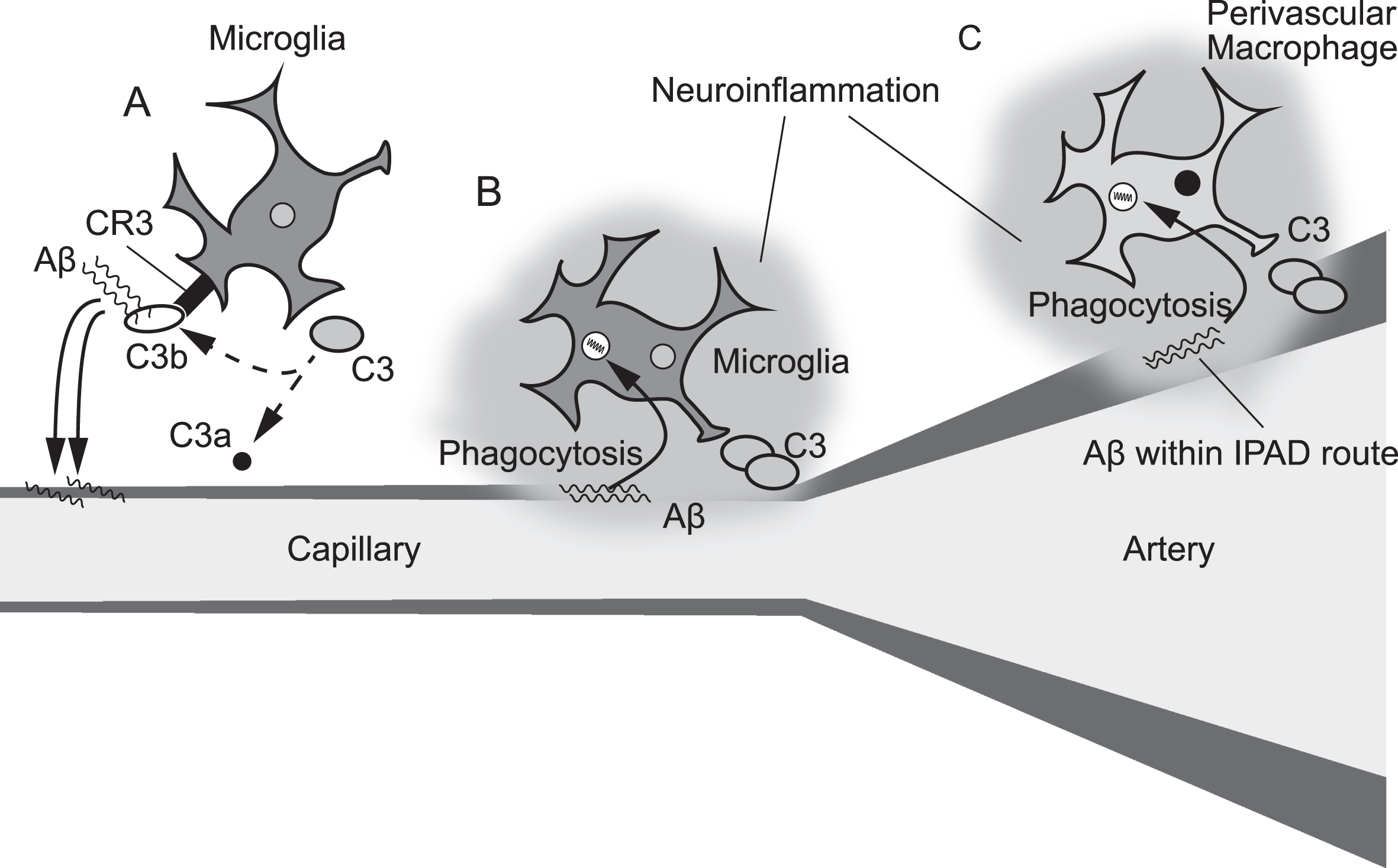
Three distinct hypothetical models of the mechanism contributing to elevated C3 levels in cerebral amyloid angiopathy. Complement 3 (C3) is expressed in both microglia and perivascular macrophages in the brain. Cleavage of C3 generates C3a and C3b. Activated microglia express the complement receptor CR3 and can thus bind the C3b-amyloid-β (Aβ) complex. Microglia may relocate the C3b-Aβ complex from the brain parenchyma to the vessel wall (A). Microglia around the capillary cerebral amyloid angiopathy deposits can engulf Aβ, which may start a cascade of neuroinflammation (B). Perivascular macrophages may take up Aβ within the intramural periarterial drainage (IPAD) pathways (C).
The modified Boston criteria have been widely applied for CAA diagnosis [20]. However, its associated imaging features such as CMBs or siderosis are irreversible markers of the disease [27]. Blood biomarkers are of interest because they can be sampled repeatedly, allow for measurement of a variety of different disease-related processes, and provide insights into disease dynamics. Although several molecules have been reported as markers associated with CAA [27–30], no blood tests are currently established for CAA diagnosis. Our findings suggest that C3 may be a useful diagnostic biomarker for CAA.
A strength of this study is that patients with early-phase CAA were examined. To reduce the confounding effects of concomitant AD pathology, dementia patients were excluded. Subsequently, the activity of daily living was preserved in our cohort. There are also some limitations of our study. First, this was a retrospective study performed at a comprehensive stroke center, which introduces a risk of selection bias. Second, the limited number of cases may have affected the results. Third, only one blood sample was taken from each patient. Thus, our findings should be verified by large-scale multicenter prospective studies.
In conclusion, blood levels of C3 were increased in CAA patients. Thus, chronic inflammation including complement activation may play a pivotal role in CAA.
Footnotes
ACKNOWLEDGMENTS
This study was funded by a Grant-in-Aid from the Japan Society for the Promotion of Science Fellows (19J00106) and a Grant-in-Aid for Young Scientists (21K16944). We thank Ms. Yuko Kiyama and Ms. Natsuki Hanada for technical assistance, Dr. Kazuhiko Uchida and Dr. Hideaki Suzuki at Molecular and Clinical Bioinformatics Inc. for insightful discussion, and Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript. We used samples acquired from the National Center Biobank Network (NCBN)/ NCVC Biobank resource. For further details see http://www.ncbiobank.org/ and ![]() .
.