
Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disorder worldwide. Although the main cause of the onset and development of AD is not known yet, neuronal death due to pathologic changes such as amyloid-β (Aβ) deposition, tau aggregation, neuroinflammation, oxidative stress, and calcium dyshomeostasis are considered to be the main cause. At the present, there is no cure for this insidious disorder. However, accurate identification of molecular changes in AD can help provide new therapeutic goals. Caspases are a group of proteases which are known because of their role in cellular apoptosis. In addition, different caspases are involved in other cellular responses to the environment, such as induction of inflammation. Emerging evidence suggest that these proteases play a central role in AD pathophysiology due to their role in the processing of amyloid-β protein precursor, tau cleavage, and neuroinflammation. Therefore, it seems that targeting caspases may be a suitable therapeutic option to slow the progression of AD. This review focuses on the role of caspases in AD pathophysiology and introduce results from studies targeted caspases in different models of AD.
BACKGROUND
Alzheimer’s disease (AD), caused by loss of neurons and synapses in central nervous system, is the most common neurodegenerative disorder worldwide making up 60% –80% of all dementia cases and affecting an estimated 24 million people globally [1, 2]. Although AD can occur in young persons, it is known as a disease of elderly, in a way that the AD prevalence increases significantly with advancing age, with a greater than 15-fold increase observed between the ages of 65 and 85 [3]. AD is characterized by a progressive decline in cognition affecting multiple systems, including language, memory, and executive functions [4]. In recent years, hypoxia resulted by cerebral perfusion alterations has been proposed to be the leading factor the pathogenesis of early stages of AD [5, 6]. However, deposition of extracellular amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs) are considered as the main molecular changes in AD brains [7]. Increasing evidence suggest that Aβ plaques and NFTs in AD brains contribute to neuroinflammation; however, in recent years, macrophage dysfunction following gene mutations has been suggested as another cause of neuroinflammation in AD [1]. Oxidative stress, caused by mitochondria alterations, and calcium dyshomeostasis are the other pathologic changes in AD, which seem to be the leading cause of apoptosis induction and neuronal loss [7, 8]. In this regard, caspases are the most important pro-apoptotic factors, which their aberrant activity has been detected in AD brains. In addition to apoptosis, it is clearly understood that caspases play great parts in amyloid-β protein precursor (AβPP) processing, Aβ production, tau cleavage, and neuroinflammation. On the other hand, Aβ deposition and NFTs has been introduced to activate caspases by different mechanisms, leading to induction of neuronal apoptosis. This paper aims to review the role of caspases in different aspects of AD pathophysiology and consequences of therapeutic interventions to modulate their activity in AD.
The caspase family of proteases
The first discovery of the mammalian caspases and their roles in regulation of apoptosis returns to identification of the requirement of the Caenorhabditis elegans gene Ced3 for apoptosis in the nematode [9]. Clonization and sequencing of the Ced3 gene product contributed to determination of a homologous relationship between this product and mammalian cysteine protease caspase-1, which was then called interleukin-1β converting enzyme (ICE) [10]. Similar homology was found between cell death protein 3 (CED-3) and multiple additional mammalian proteins which were then known as the caspases [11]. The name ‘caspase’ comes from an active site cysteine which prefers (‘C’), and target substrates cleavage after an aspartate residue that prefers (‘asp-ase’) [11]. So far, 14 mammal caspases and 12 human caspases are known. However, these proteases are zymogens consisting of three domains, including the pro-domain and the large and small catalytic domains. In order to activate caspases, cleavage between these domains is necessary, leading to the generation of a heterotetramer active caspase with two large and two small domains [12]. Although each caspase has its specific substrate, they all affect the aspartate residue prior to the cleavage site [13]. It has also been observed in some cases that caspases can perform cleavage after phosphoserine and glutamate residues [14]. Caspases are divided into two subgroups, including initiator (i.e., caspase-2, -9, -8, and -10) and executioner caspases (i.e., caspase-3, -6, and -7). The main difference between these two subgroups is due to the presence of long pro-domains comprised of protein:protein interaction motifs in initiator caspases. In addition, differences between caspases in each subgroup returns to differences in their catalytic domains (reviewed in [15]). However, interaction domains in initiator caspases play a role in the recruitment of these caspases multi-protein complexes which act as activators for other caspases. These activators are specified for each caspase, which include death inducing signaling complex (DISC) for caspase-8 and -10, p53-induced death domain protein (PIDD) apoptosome for caspase-2, and apoptotic peptidase activating factor 1 (APAF1) apoptosome for caspase-9 [15]. In addition to mode of activation and structural differences, initiator and executioner subgroups of caspases are separated due to their substrate preferences. For instance, positional scanning peptide libraries has determined that caspase-3 and -7 have similar substrate sequences [13]. Figure 1 depicts the classification of caspases based on their structure and function.
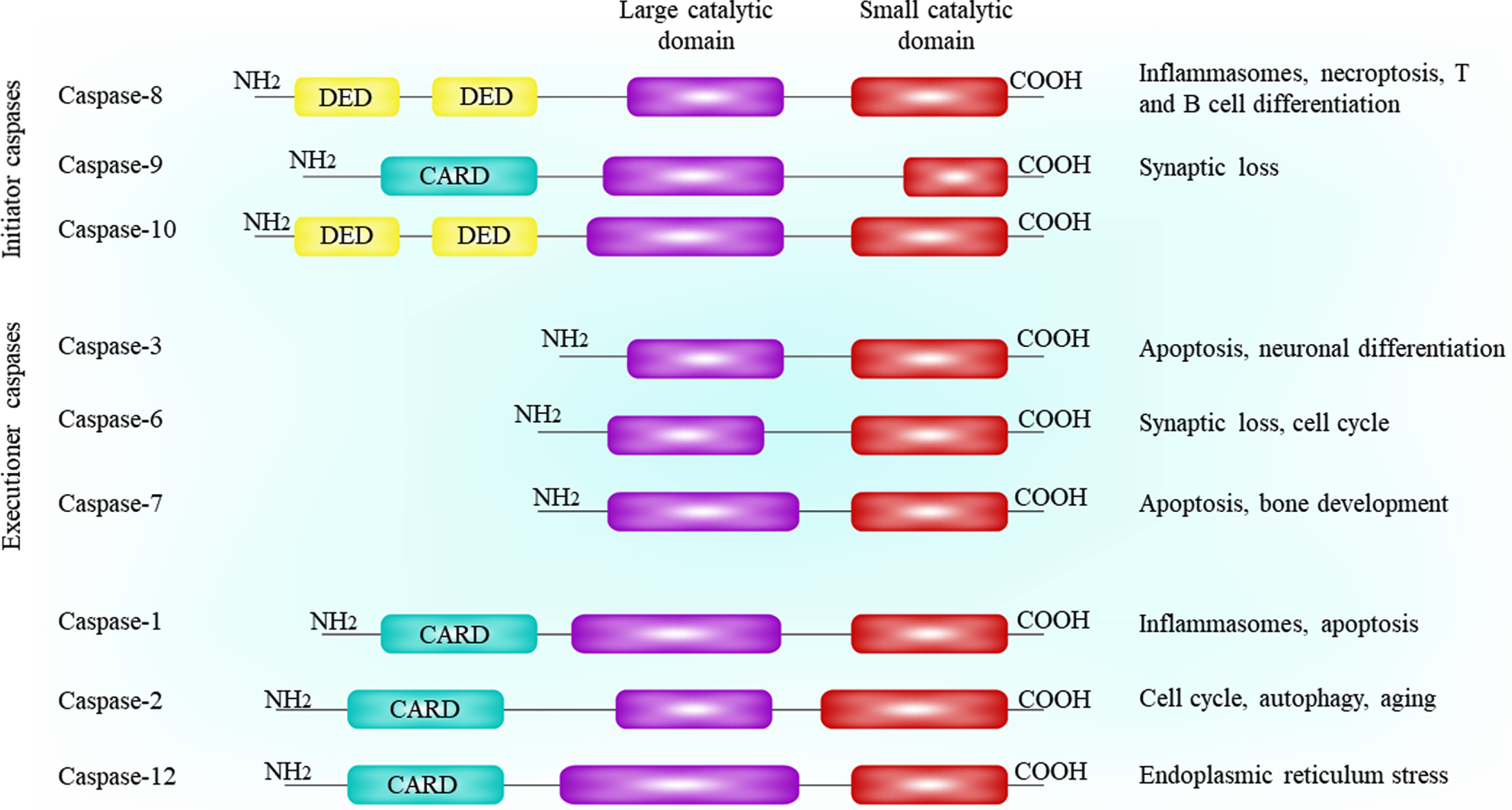
The members of caspase family and their functional and architecture classification.
Activation of caspases
The crystal structures of the catalytic domains of different caspases exhibit a homodimer stable structure in the catalytic domains. An active site consists of 5 loops (L1–4 and LH) represented on either end of the dimer of each homodimer. Executioner caspases are present as preformed dimers in the cell which are activated by cleavage [16]. Upstream caspases or other proteases such as granzyme B are involved in activation of executioner caspases which act through cleavage of the linker between the large and small subunits [17]. In the uncleaved form of caspase, the active site is occupied, which blocks the binding of a substrate. In caspase-3 and -7, this occupation occurs by the inter-domain linker [18–20].
Contrary to executioner caspases, initiator caspases are activated through a dimerization process. Followed by dimerization, the active site is created. Activation of caspase-9 can occur without cleavage, and cleavage block contributes to loss of 80% of caspase activity [21, 22]. It has been demonstrated that caspase-8 and -9 enforced dimerization exhibits an enhanced activity. In addition, it has been shown that blocking the cleavage of caspase-8 leads to impair the dimeric species stability [16].
Caspase signaling in apoptosis
There are two main pathways to activate caspases in induction of cell apoptosis including intrinsic and the extrinsic pathways [23]. The separation of these pathways is due to stresses that come from outside the cells or within the cell to activate apoptosis cascade. Intracellular stresses which contribute to activate the intrinsic pathway include metabolic stress, DNA damage, and endoplasmic reticulum stress [24]. These stresses affect mitochondria and contribute to cytochrome c release from the mitochondrial intermembrane space to the cytosol followed by mitochondrial outer membrane permeabilization. In the cytosol, cytochrome c binds to APAF1 through its WD repeats leading to the opening up of the molecule followed by a conformational change. Presence of dATP or ATP contributes to APAF1 oligomerization and APAF1 apoptosome formation [25, 26]. At the next step, the apoptosome activates caspase-9 which continues the cleavage of the caspases-3 and -7. Caspase-9 tethered to the apoptosome is required for caspase-3 cleavage and, also, caspase-9 auto-cleavage and dissociation from the apoptosome [27].
At the extrinsic pathway of apoptosis, binding of a death ligand to a death receptor (i.e., tumor necrosis factor receptor) leads to activation of apoptosis cascade [28]. The death receptors have a death domain (DD) at their C-terminal which shares structural features with two domains of initiator caspases, death effector domain (DED) and caspase recruitment domain (CARD). Each of the CD95 and TNF-related apoptosis-inducing ligand (TRAIL) receptors, as the main death receptors, are activated followed by multimerization due to binding of their cognate death ligand leading to activate platform assembly and caspase-8. Assembly of each TRAIL and CD95 leads to DISC formation at the plasma membrane. Regarding the TNFR1, activation of caspase-8 platform does not initiate due to assembly at the membrane. In this regard, TNFR1-TNF axis rapidly recruits receptor-interacting serine/threonine-protein kinase 1 (RIPK1), a Fas-associated protein with death domain (FADD)-like homolog, and TNF receptor associated factor 2 (TRAF2) at the plasma membrane [29]. This process leads to the generation of two complexes namely complex I and complex IIa. Complex I activates nuclear factor kappa B (NF-κB) and complex IIa recruits caspase-8 to induce apoptosis [29]. In the absence of caspase-8, complex IIa induces another form of cell death which is called necroptosis, followed by activation of mixed lineage kinase domain like pseudo-kinase (MLKL), involved in pore formation in the membrane [30].
The recruitment of caspase-8 to the TNFR complex II or DISC occurs by adaptor molecule FADD. DED pro-domains of caspase-8 form filaments followed by recruitment to the DISC, which leads to its activation [31]. The first caspase-8 DED binds to FADD and the second one binds to the first DED of another caspase-8 by tandem linkage, which provides the mechanism of initiation the caspase-8 cascade [32, 33]. The extrinsic and intrinsic pathways of apoptosis are represented in Fig. 2.
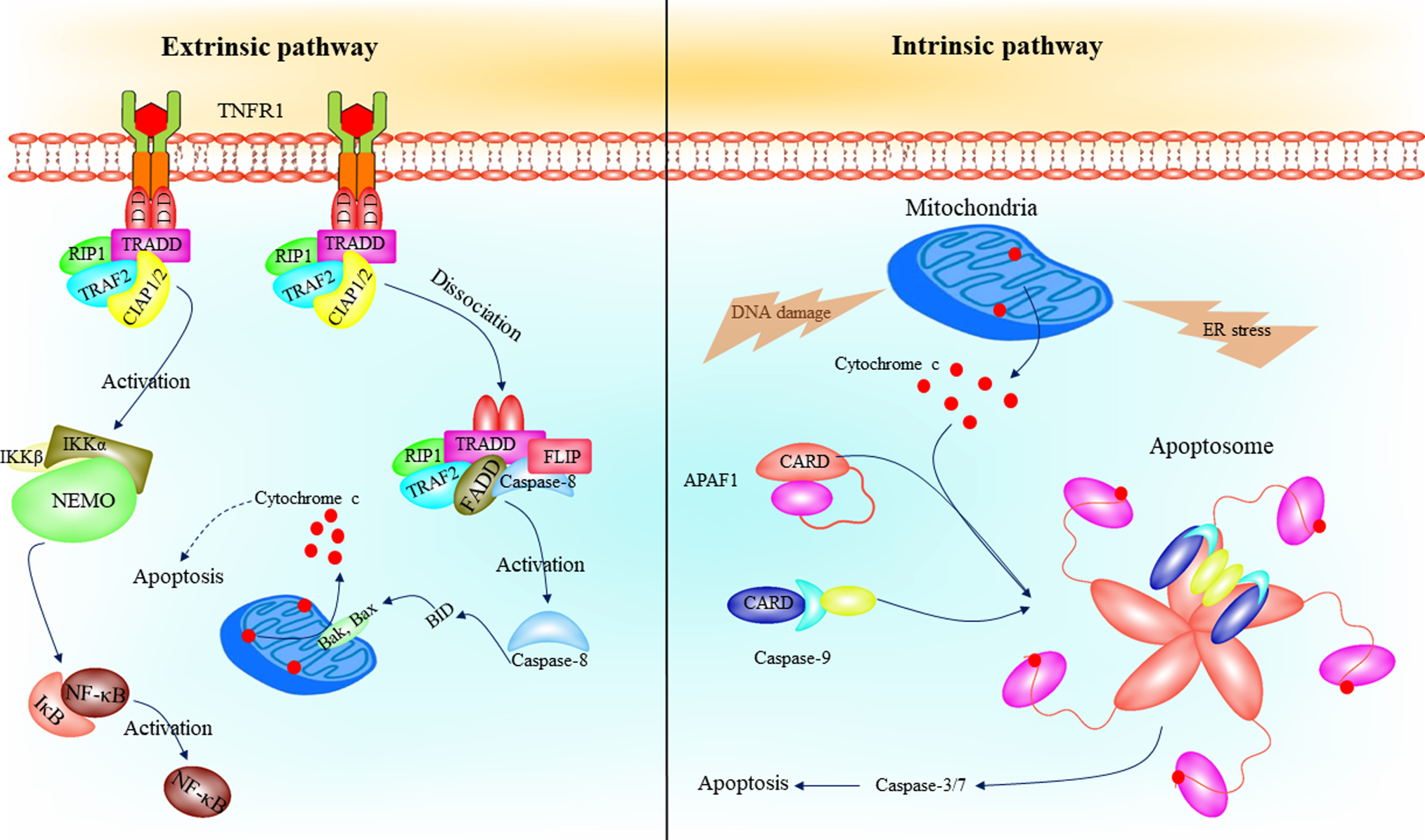
The intrinsic and extrinsic pathways of apoptosis. Several intracellular stresses, such as endoplasmic reticulum and DNA damage, contributes to MOMP and intrinsic apoptosis pathway initiation. Cytochrome c release followed by MOMP, which binds to the WD repeats (pink ovals) of the APAF1 leading to apoptosome formation. The apoptosome activates caspase-9, and eventually, activates caspase-3 and -7 leading to apoptosis. In the extrinsic pathway of apoptosis, binding of a death ligand, such as TNF-α, to a death receptor contributes to caspase-8 activation followed by DISC formation. Caspase-8 activates BID, which leads to Bak and Bax oligomerization and cytochrome c release from mitochondria. On the other hand, TNF-TNFR axis contributes to activation of NF-κB by mediating of IKK complex phosphorylation. APAF1, apoptotic peptidase activating factor 1; DISK, death-inducing signaling complex; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; TNF-α, tumor necrosis factor α; IKK, inhibitor of NF-κB kinase.
In addition to intrinsic and extrinsic pathways of apoptosis, there is another mechanism for apoptosis induction which occurs by caspase-2. This caspase cleaves the BH3-interacting domain death agonist (BID) and pro-apoptotic B-cell lymphoma 2 (BCL2) family protein leading to mitochondrial outer membrane permeabilization and activation of caspase-3 and -9 [34].
CASPASES AND ALZHEIMER’S DISEASE
Although the underlying cause of AD is not yet known, Aβ plaque accumulation and tau hyperphosphorylation and aggregation, generating NFTs, are known to be the main characterizations of the disease [1]. However, whatever the cause of AD, it seems that eventually causes neuroinflammation and oxidative stress, leading to neuronal death [35]. In different studies, it has been shown that pro-apoptotic factors have been activated and contribute to induction of apoptosis of neurons. Caspases, as the main mediators of cellular apoptosis, play a crucial role in different aspects pathophysiology of AD. On the other hand, increasing evidence indicate altered expression or activity of caspases in different samples from AD patients. Table 1 summarizes changes in the levels of different caspases in samples from patients with AD. Here we review the role of caspases as the main inducers of apoptosis in different aspects of AD pathophysiology.
Dysregulated caspase activity in samples from AD patients
Caspases and AβPP processing in Alzheimer’s disease
In AD brains, it seems that there is an aberrant production and clearance of Aβ leading to its deposition [36]. In addition, amyloidogenesis and subsequent Aβ accumulation has been associated with different pathologies [37]. However, Aβ is produced from AβPP, which is highly expressed in neurons. There are two pathways for AβPP processing termed amyloidogenic and non-amyloidogenic pathways. In the amyloidogenic pathway, firstly, AβPP is cleaved by β-site amyloid precursor protein cleaving enzyme 1 (BACE-1), one of the main β-secretases leading to generate and release β-stubs. In addition, cleavage of transmembrane AβPP by BACE-1 leads to liberate soluble N-terminus of AβPP, while the C-terminal fragment (C99) is still attached to the membrane. At the second step, cleavage of C99 occurs by γ-secretase which contributes to release the final Aβ form to the extracellular space [38]. Non-amyloidogenic pathway is initiated by cleavage of AβPP by α-secretase leading to release soluble AβPPα [39].
These processes show the presence of a wide range of proteases. It is clearly understood that caspases as a group of intracellular proteases are involved in AβPP processing. It was first established that caspases are involved in synaptic injury in AD [40–43]. It has been reported that caspase-3 activates calcineurin leading to removal of GluR1 from postsynaptic sites in AβPP transgenic mice and subsequently, behavioral impairment followed by spine degeneration [40]. To determine the role of caspases in synaptic alterations in AD, a study designed by Park et al. [44] showed that Aβ-induced synaptic injury is mediated by activation of caspases leading to induce AβPP processing. In a closer inspection, it has been indicated that initiator caspases-8 and -9 activation by Aβ leads to cleave AβPP and generation of a C31 fragment, which acts as a cytotoxic factor and causes synaptic loss [45]. In addition to the mentioned points, it has been demonstrated that caspases cleave AβPP directly, leading to generate Aβ [46]. In this study, it was reported that neuronal NT2 cells generate elevated levels of Aβ during apoptosis which is attenuated by caspase inhibitors. Additionally, it was shown that caspase-3 is the main caspase involved in AβPP processing to produce Aβ, which cleaves AβPP at Asp720. However, regarding the mechanism of caspase activation by Aβ, it has been proposed that Aβ binding contributes to promote AβPP dimerization and formation of a cleavage site after D 664 by caspases leading to generate C31 peptide [47]. The important point about these studies is that Aβ accumulation is not seen in the early stages of AD [1], therefore, it is not clear that caspases are activated in the early stages of the disease and cause synaptic disorders. If the activity of caspases is increased in the early stages of AD leading to synaptic loss, what factor causes this increase in activity?
In answer to these questions, hypotheses other than the role of Aβ in caspase activation can be proposed. In this regard, it can be referred to intracellular pathways, the phosphatidylinositol 3 kinase (PI3K)/AKT and extracellular signal-regulated protein kinase (ERK)1/2. These pathways are involved in regulation of different aspects of cell life, including protein synthesis and regulation of apoptosis [48, 49]. They regulate cell apoptosis through inhibition of pro-apoptotic factors, especially caspases, and activation of anti-apoptotic factors [50, 51]. In addition, activation of both of these pathways has been shown to have protective effects against synaptic impairment [52, 53]. On the other hand, reduced activity of the PI3K/AKT and ERK1/2 pathways has been shown in AD brains, which can lead to over-activity of caspases [1, 54]. However, further studies are required to clarify the role of these pathways in synaptic loss though caspases in early stages of AD. In addition to mentioned points, interaction between the PI3K/AKT pathway and Aβ may be another mechanism for caspase activation in AD, as it has been shown that Aβ oligomers suppress this pathway leading to induce apoptosis [55, 56].
Mechanism of Aβ-induced caspase activation
In addition to the role of different caspases in AβPP processing, numerous studies indicate the role of Aβ in activation of intrinsic and extrinsic pathways of apoptosis. To investigate the effect of Aβ on the activation of the intrinsic pathway of apoptosis, its entry into neurons must first be examined. Different routes for Aβ entry to neurons has been introduced, the most important of which are several receptors including acetylcholine and glutamate receptors, receptor for advanced glycation end products (RAGE), and serpin-enzyme complex receptor (SEC-R). In addition to receptor-mediated entry of Aβ, receptor-independent mechanisms have been introduced as ways for Aβ entry to the neuron, such as clathrin-mediated endocytosis [57]. However, Aβ entry to the neurons contributes to release of cytochrome c from mitochondria resulted by mitochondrial permeability transition pore (MPTP) opening, leading to Apaf-1 formation and continuous apoptosis cascade as described in previous parts [58]. Regarding the extrinsic pathway of apoptosis, it has been proposed that AβPP or Aβ bind to death receptor 6 leading to recruitment of FADD and caspase-8 which contributes to formation of DISC complex and activation of caspase-3 and -6 [59].
Aβ-induced caspase activation: Indirect mechanisms
In addition to direct activation of caspases by Aβ, several indirect mechanisms can be involved. One of the most important consequences of Aβ deposition is calcium dysregulation in neurons. Interactions between Aβ oligomers and several calcium channels have been introduced as the main mechanism for increase intracellular calcium and its dysregulation. In this regard, several studies indicated that Aβ oligomers are able to increase calcium influx via N-Methyl-D-aspartate (NMDA) receptors. This process has been proved by memantine, an uncompetitive NMDA receptor antagonist, which augmented Aβ oligomer-induced calcium increase and toxicity [60]. In addition, Aβ induces dynamin 1 degradation, affecting synaptic integrity, which is mediated by activation NMDA receptors [61]. In addition to NMDA receptors, several other receptors have been identified mediating Aβ-induced calcium dysregulation, such as voltage-gated Ca2 + channels [62], catecholamine [63], nicotinic acetylcholine [64], and serotonin receptors [65]. Additionally, formation of calcium-permeable pores at the cell membrane by Aβ is the other known mechanism for calcium dysregulation [66]. However, increased intracellular calcium followed by Aβ-induced toxicity contributes to activation of calpain, which eventually leads to activation of caspase-12 [67]. At the next step, caspase-12 induces caspase-9 activation which activates caspase-3 and -7 [68].
In addition to dysregulation of cellular calcium, Aβ causes mitochondrial calcium dyshomeostasis. It has been demonstrated that accumulation of both AβPP and Aβ in mitochondrial matrix occurs through translocase of the outer mitochondrial membrane (TOM) 40 and translocase of the inner mitochondrial membrane (TIM) 23 import channels [69, 70]. This process has been proved by detection of Aβ plaques in mitochondria of human postmortem AD brains [71]. However, Aβ entry to the mitochondrial matrix leads to impair mitochondrial calcium and consequent activation of MPTP, which itself contributes to release cytochrome c and induction of caspase cascade [72, 73]. On the other hand, it has been proposed that interaction between Aβ and an MPTP regulator, cyclophilin D, leads to increase reactive oxygen species (ROS) production and oxidative stress induction [74], which may be another mechanism for activation of caspases [75].
Caspases and tau in Alzheimer’s disease
The microtubule-associated tau protein plays a great part in stability of axonal microtubules and regulation of axonal transport [76]. However, the association between tau and AD is related to formation of NFTs, a pathological characterization found in AD brains [77]. NFTs are neurotoxic aggregated proteins which have been associated with synaptic alterations, mitochondrial dysfunction, and oxidative stress leading to loss of neurons in the brain of AD patients [78–80]. There are two main tau modifications observed in AD models, including tau hyperphosphorylation and tau aggregation [81]. However, regarding the association between caspases and tau in AD, both seem to affect each other and there is an interrelationship between them. Caspases are involved in tau aggregation, but not its hyperphosphorylation in AD, and caspase-cleaved tau has been detected in AD brains [81]. In addition, caspase-cleaved tau at D421 has been shown to be an early event in AD tangle pathology [78]. It seems that two caspases, caspse-2 and -3, are the most important ones in tau cleavage in AD. It has been demonstrated that caspase-2 cleaves tau at Asp314 leading to generate a soluble tau fragment which has been associated with dementia progression [82]. Tau cleavage at D421 by caspase-3 has been introduced as one of the main inducers of NFTs formation has been observed in AD [83, 84]. In particular, caspase-3-cleaved tau at D421 in in aged mice has been found to be associated with synaptic plasticity deficits and cognitive impairment [85]. In a closer inspection, another study indicates that caspase-3 is involved in cleavage of both N- and C-terminals of tau, in a way that caspase-3 inhibition by Z-VAD-fmk reduces its cleavage activity on both terminals [80]. Also, caspase-cleaved tau has been shown to exhibit a rapid memory impairment in TauC3 mouse as an AD model [86]. However, in transgenic knock-in mice expressing full-length human tau 0N4R only or together with human caspase-6, it has been indicated that caspase-6-cleaved tau at D402 fails to cause tau hyperphosphorylation and aggregation, neuroinflammation, and neurodegeneration [87].
The other aspect of consideration of tau-caspase association is the role of tau in caspase activation as shown in several studies [88]. In this regard, it can be re-referred to calcium dyshomeostasis and oxidative stress induced by tau. It has been indicated that treatment of primary cortical cultures with tau and iPSC-derived neurons with mutations in MAPT encoding tau contributes to suppress mitochondrial calcium efflux through inhibition of mitochondrial Na+/Ca2 + exchanger [89]. In this study, it was observed that elevated calcium followed by tau toxicity leads to induce neuronal apoptosis by mediating of caspase-3 activation. In another study, caspase 3-cleaved tau at the C-terminus was observed in outer mitochondrial membrane and within the inner mitochondrial space, suggesting a regulatory effect for tau on mitochondrial function [90]. Also, in this study it was shown that caspase 3-cleaved tau alters endoplasmic reticulum calcium homeostasis. In addition to these studies, it has been demonstrated that caspase-3-cleaved tau induces mitochondrial fragmentation, ATP loss, and increase ROS generation, which can be another way for caspase activation [91]. Figure 3 represents the role of caspases in the pathophysiology of AD.
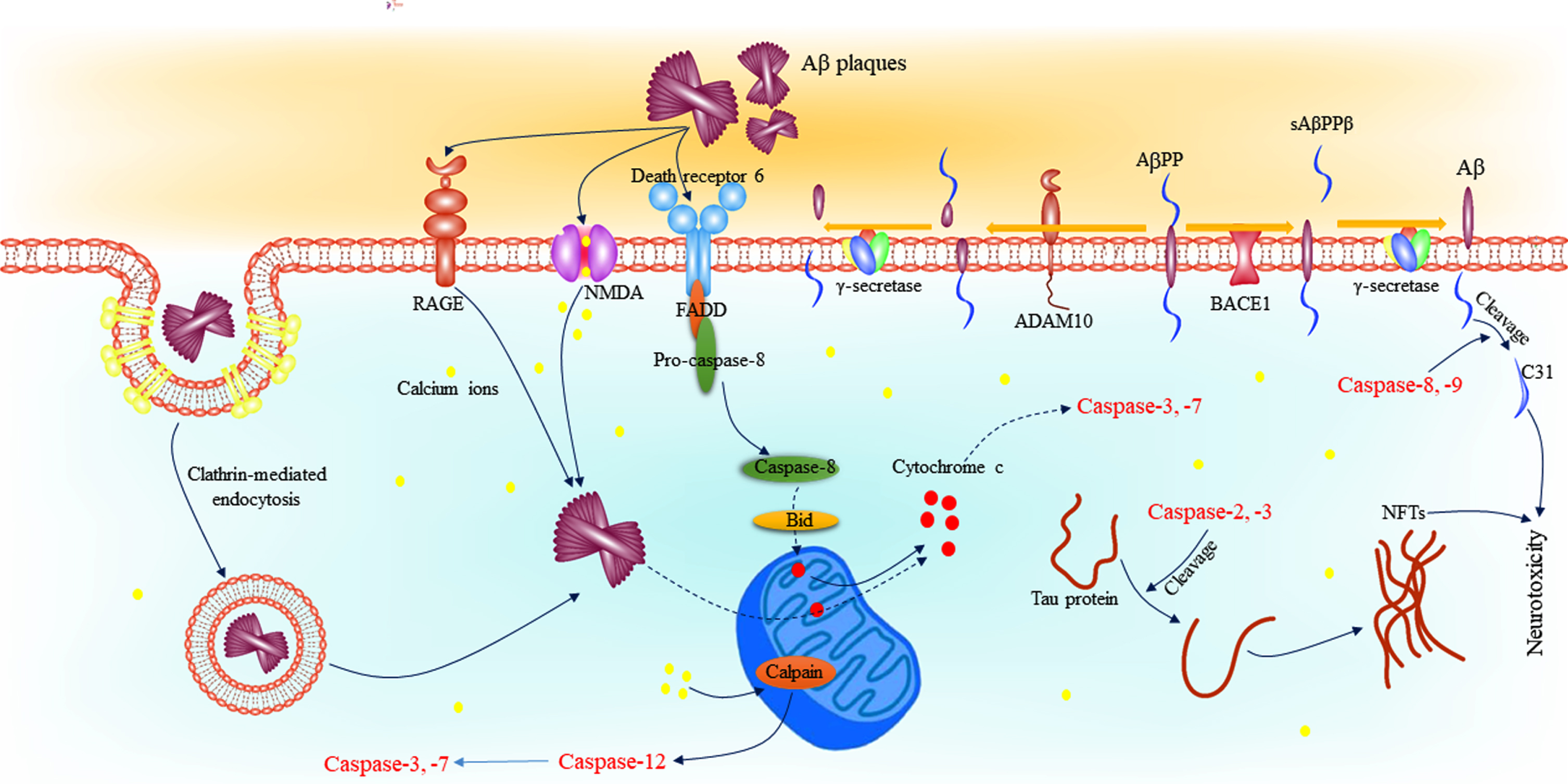
Role of caspases in the pathophysiology of AD. Caspase-8 and -9 are involved in the cleavage of AβPP leading to formation of C31, and fragment involved in synaptic loss in AD. In addition, caspase-2 and -3 cleave tau proteins which contributes to formation of NFTs. Binding of Aβ plaques to death receptor 6 recruits FADD and converts pro-caspase-8 to its active form. Activated caspase-8 activates BID leading to cytochrome c release and caspase-3 and -7 activation. On the other hand, Aβ plaques are accumulated in the cell through NMDA, RAGE, and clathrin-mediated endocytosis leading to release cytochrome c. Also, the effect of Aβ plaques on NMDA receptors and mitochondria leads to dysregulation of intracellular calcium. Increased calcium levels into the cell activates calpain, and eventually, caspase-3 and -7 by mediating of caspase-12. Aβ, amyloid-β; AD, Alzheimer’s disease; AβPP, amyloid-β protein precursor; FADD, Fas-associated protein with death domain; NFT, neurofibrillary tangles; NMDA, N-methyl-D-aspartate.
Caspases and neuroinflammation in Alzheimer’s disease
Increasing evidence indicate that AD pathogenesis is not limited to neuronal compartment, but includes interactions between aggregated proteins and several recognition receptors on microglia, as the main residents of neuroinflammation in neurodegenerative diseases [35, 92]. However, detection of AD-specific microglia with different gene expressions [93] and several gene mutations, such as TREM2 and CD33 [94], in AD brains indicate mechanisms for microglia activity in AD rather than their activation by aggregated proteins. Activation of microglia cells by Aβ plaques, NFTs, or any other reason contributes to quick release of inflammatory cytokines, as observed in different samples from AD patients (reviewed at [95]). Several evidence suggest a regulatory effect for caspases in microglial activation [96]. Stimulation of BV2 microglia cells by lipopolysaccharide (LPS) has been shown to induce caspase-3 cleavage and DEVD-ase (caspase-3/7) activity [97]. In this study, increased DEVD-ase activity was also observed followed by stimulation of toll-like receptor (TLR)2 by PamC3sk4, a synthetic lipopeptide, and lipoteichoic acid. Although caspase-3 and -7 are known as pro-apoptotic factors, their activation in this study did not lead to microglial apoptosis. Interestingly, it was shown that DEVD-ase is associated with IKK-b expression leading to increase IkB phosphorylation and ubiquitination which eventually contributes to NF-κB activation. This process proved by treatment of caspase-3 and caspase-7 knockout microglia by LPS which associated with loss of microglia neurotoxicity. On the other hand, activation of caspase-3 and -7 was shown to be mediated by caspase-8 activation through TLR4, but not a FADD-dependent mechanism. Also, interaction between caspase-3 and -7 with IKK/NF-kB pathway followed by LPS-induced microglia activation was shown to be mediated by caspase-cleaved protein kinase C (PKC) leading to generate its 40-kDa active fragment. Although the effects of Aβ on caspase-mediated microglia activity were not investigated in this study, but the activation of TLR4 by Aβ oligomers [98] could indicate their role in this process. However, the role of caspase-1 has been investigated in neuroinflammation in J20 mouse model of AD [99]. In this study, it was shown that treatment of J20-treated mice with VX-765, a selective caspase-1 inhibitor, restores decreased type I resting microglia and increased type II and III activated microglia. In addition, treatment with VX-765 contributed to reduce the expression of pro-inflammatory cytokines, such as interleukin (IL)-1 and tumor necrosis factor α (TNF-α).
In addition to mentioned processes, caspases are involved in regulation of other microglial activities. NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3), or cryopyrin, is an intracellular complex involved in microglia response to Aβ plaques [98]. NLRP3 serves as a sensor molecule which together with pro-caspase-1 and adaptor protein ASC forms active NLRP3 inflammasome. Elevated levels of NLRP3 inflammasome has been detected in AD brains [100], and its activation has been associated with tau pathology induction [101], which leads to introduce it as a therapeutic target for AD [102]. Activation of NLRP3 inflammasome contributes to activate caspase-1 which eventually cleaves IL-18 and IL-1β and converts them to an active form [103]. Increased IL-1β has been proposed to induce glycogen synthase kinase 3 (GSK-3) activation, tau hyperphosphorylation, and NFTs formation through activation of IL-1β receptor [104]. However, although caspase-1 activity in NLRP3 inflammasome has been well known, the role of other caspases in its activation is not known yet. In addition to TLR4-mediated NLRP3 inflammasome activation, triggering receptor expressed on myeloid cells 2 (TREM2) is involved in regulation of NLRP3 inflammasome and caspase-1 activity followed by interaction with Aβ plaques [105]. Also, activation of TREM2 by Aβ contributes to inhibit microglial apoptosis through activation of the PI3K/AKT pathways and caspase-3 inhibition [105].
The other aspect of considering the association between neuroinflammation and caspases relates to their activation followed by pro-inflammatory effects on targeted neurons [106]. For instance, binding of TNF-α to TNF receptor 1 (TNFR-1) contributes to recruitment of adaptor proteins such as RIP1, TRADD, TRAF2, pro-caspase-8, and FADD to form complex II [107]. In activated complex II, FADD induces caspase-8 activation leading to trigger caspase cascades and apoptosis induction [29].
Caspases and autophagy in Alzheimer’s disease
Autophagy is known as a conserved catabolic process involved in degradation of defective organelles or proteins in lysosomes and recycling basic components in eukaryotic cells [108]. In recent years, the association between autophagy and AD has been investigated in numerous studies since autophagy plays a central role in clearance of insoluble protein aggregates in mammalian nervous system to maintain protein homeostasis [109]. Increasing evidence has shown that altered autophagy leads to AD pathogenesis. Initially, electron microscopy analysis elucidated that autolysosomes and autophagosomes accumulate in the brains of AD patients [110]. However, other investigations clarified more facts about the association between autophagy and AD pathology. It is clearly understood that autophagy plays a central role in Aβ metabolism mainly via regulation of its generation and clearance. In this case, it has been shown that autophagy is involved in AβPP degradation [111], Aβ secretion [112], and its clearance [113]. Autophagy also participates in tau pathology other than Aβ metabolism, as it has been observed that blockage of autophagic flux accompanied with accumulation of insoluble tau aggregates [114]. In addition, in familial AD brains, colonized hyperphosphorylated tau with the autophagy receptor p62/SQSTM1 and autophagosome marker LC3 was observed, while this overlap was not found in healthy controls [115]. In addition to tau aggregation and Aβ deposition, accumulating evidence introduce functional autophagy as a crucial factor in synaptic functions, mainly synaptic plasticity and neurotransmission [116].
The impact of different caspases in autophagy process has been investigated in numerous studies. Initially, it was clearly understood that caspases are involved in cleavage of many autophagy-related proteins (ATGs) [117–119]. Caspases-ATG protein interaction results in abolition of the autophagic function of the latter that way the homeostatic balance towards the apoptotic profile [120, 121]. In addition, caspases have been also shown to be involved in autophagy promotion under certain conditions [122, 123]. Here we briefly review the association between caspases activity and autophagy. In casp-1–/– mice hepatocytes, a decrement in LC3 and Beclin-1 as autophagy markers was detected compared to wild-type hepatocytes, suggesting the role of caspase-1 in clearance of redox stress-induced damaged mitochondria, possibly via activating LC3 and Beclin-1 [124]. The results of several studies in mouse embryonic fibroblasts [125] and young adult mouse cortical neurons [126] indicate that caspase-2 regulates autophagy negatively. Pro-caspase-8 can also be involved in regulation of autophagy process, as its localization to the autophagosomes has been observed via p62, an ATG8/LC3-interacting molecule binding poly-ubiquitinated protein aggregates for their sequestration at the autophagosome formation site [127, 128]. In addition, activated caspase-8 has been elucidated to inhibit overactivation of autophagy [129]. Regarding caspase-9, it has been reported that this caspase inactivates ATG5 and Beclin-1 via their cleavage in certain sites [130, 131]. In addition to the mentioned points, the association between effector caspases and autophagy process has been investigated in several studies. In this regard, it has been reported that proteolytic cleavage of Beclin-1 by caspase-3 is sufficient in impairing autophagic flux followed by application of an apoptotic-inducing stimulus [130]. In addition, ATG5 has been proposed to be a target for caspase-3-mediated cleavage consequently leading to autophagy downregulation [131]. In regards to the interaction of caspase-6 with autophagic proteins, it has been reported that this caspase exacerbates Beclin-1 and ATG5 degradation leading to inhibiting autophagy [131].
The association between caspases activity and autophagy in AD is the other issue which should be examined. Crosstalk between caspase-3 induced neuronal apoptosis and autophagy has been reported in aging PS/APP mice [132]. In another study, depletion of Beclin-1 was detected in AD brains which was linked to its cleavage by caspase-3 leading to formation of a 35-kDa C-terminal fragment [133]. Although the regulatory role of caspases in regulating the autophagy process is clear, this association has been less investigated in AD models. In this context, the effect of various caspases on Aβ, tau protein, and oxidative stress in AD models by mediating of autophagy modulation can be investigated.
microRNAs as caspase regulators in Alzheimer’s disease
miRs are a group of non-coding RNA molecules with approximately 25 nucleotides. microRNAs regulate the expression of a wide range of proteins through binding to their 3’UTR mRNA [134]. However, the role of miRs in different pathologies has been investigated in recent years. It is clearly understood that alterations in miR levels, which has been detected in different samples from AD patients, is strongly associated with pathophysiology of disease [135]. miRs has been shown to be involved in regulation of Aβ production and clearance, NFT formation, neuroinflammation, and neuronal apoptosis [135]. The association between miRs and neuronal apoptosis in AD is because of their regulatory effect on different factors involved in apoptosis, such as caspases. In addition, miRs affect caspases to regulate several other pathological changes in AD. In this regard, it has been demonstrated that miR-132 attenuates tau-induced toxicity through direct regulation of calpain and caspase-3 and -7 [136]. In addition, it has been demonstrated that miR-34a targets caspase-2 directly, and its upregulation by inositol-requiring enzyme 1 exhibits protective effects against Aβ-induced injury in SH-SY5Y cells [137].
Indirect regulation of upstream pathways by miRs is the other way to regulate caspase activity in AD, as shown in different studies. For instance, it has been indicated that miR-137 suppresses caspase-3 activity through targeting TNF-α-induced protein 1 (TNFAIP1) followed by Aβ-induced neurotoxicity in Neuro2a cells [138]. Also, it has been indicated that miR-20b-5p targets Ras homolog family member C in AβPP swe/PSΔE9 mice leading to aggravate neural apoptosis through reduce cleaved-caspase-3 expression [139].
Although the role of miRs in regulating caspase activity is undeniable, most studies in AD have focused on the role of miRs in inhibiting caspase-induced apoptosis. It seems that considering the association between miRs and caspases in other aspects of AD pathophysiology, such as AβPP processing and tau cleavage, can be constructive.
Pharmacologic modulation of caspases in Alzheimer’s disease
Although AD is a public health challenge worldwide, as of now, just two classes of drugs are approved to be used in AD patients, including cholinesterase enzyme inhibitors (naturally derived, synthetic and hybrid analogues) and N-methyl d-aspartate (NMDA) antagonists. Using of acetylcholinesterase inhibitors (AChEIs) is based on the cholinergic hypothesis indicating the reduction in acetylcholine (ACh) biosynthesis in AD. On the other hand, NMDA receptor over-activation results in elevated levels of influxed Ca2 + leading to promote cell death and synaptic loss. Prevention of NMDA receptors activity by NMDA antagonists reverses these processes and has been shown to have several benefits in AD [140, 141]. However, the protective effects of mentioned drugs on several other pathologic changes in AD has been shown in several studies. For instance, it has been demonstrated that donepezil exhibits protective effects against neurodegeneration via prevention of tau pathology and anti-inflammatory properties [142]. Galantamine, another FDA-approved agent for AD, has been shown to reduce Aβ deposition and astrocyte activation in APP/PS1 transgenic mice [143]. In addition to mentioned points, several studies reveal the effect of these drugs on caspases activity in AD. In this regard, it has been reported that galantamine inhibits Aβ-induced cytostatic autophagy in PC12 cells via decreased the protein level of cleaved-caspase-3 [144]. In addition, it has been reported that treatment of PC12 cells with galantamine followed by Aβ-induced toxicity exhibits protective effects via suppressing the activity of caspase-9, caspase-12, and caspase-3 [145]. Regarding rivastigmine, in a study it was observed that rivastigmine treatment improves cognitive impairment in rats exposed to streptotozocin via inhibition of ER stress by mediating of caspase-12 [146]. Another study reveals that memantine prevents cognitive impairment in rats injected with Aβ via reduced Bcl-2 and caspase-8 activity [147].
Combination therapy has been introduced as a suitable option to increase the effect of approved AD drugs on different pathologic changes in AD. One of the most important consequences of combination therapy in AD models is increased effects of caspases activity. Using of non-steroid anti-inflammatory drugs (NSAIDs) combined with AChEIs and NMDA receptor antagonists has been shown to be one of the most effective options in this regard. For instance, it has been demonstrated that naproxen enhances anti-AD activity of rivastigmine via improvement of neurogenesis, and inhibition of caspase-mediated apoptosis leading to an additional mitigating impact on cognitive impairment [148]. In another study, it has been reported that celecoxib combined with rivastigmine confers neuroprotection via inhibition of caspase-3 activity [149]. In addition to NSAIDs, there are several other compounds used in combination with AD drugs. In this case, treatment of cultured hippocampal neurons with homocysteine and memantine followed by Aβ exposure exhibit more efficacy compared to memantine alone in downregulation of caspase-3 and -9 [150].
Increasing evidence suggest the therapeutic potential of caspase modulators in AD, and numerous studies introduce caspases as a suitable therapeutic target. In this regard, caspase inhibitors are widely used as research tools [151]. Although theoretically inhibiting caspases could have many benefits in treating of AD, or slowing its progression, no clinical studies have been performed in this regard. In non-AD pathologies, there are several studies using caspase inhibitors in clinical trials. For instance, emricasan (IDN-6556), a pan-caspase inhibitor, has been used in a phase II clinical trial on patients with liver fibrosis and nonalcoholic steatohepatitis [152]. However, although this treatment was safe and well-tolerated in these patients, it failed to exhibit proof-of-concept support for caspase inhibition as a treatment for nonalcoholic steatohepatitis and cirrhosis patients. These studies show that using of caspase-specific inhibitors in clinical trials may be a suitable option in AD patients.
The other aspect of caspase-related treatments for AD relates to natural compounds and FDA-approved drugs for non-AD diseases. These agents are not known as specific caspase inhibitors, but extensive studies have shown their effect on reducing the expression or activity of caspases in various AD models. These effects influence different aspects of AD pathophysiology, mainly Aβ and tau related pathology, and subsequent neuronal loss. Table 2 summarizes the effect of the most known drugs and natural compounds on caspases in AD models.
Therapeutic interventions to modulate the expression and activation of caspases
CONCLUSION
AD and other neurodegenerative diseases are characterized by deposition of abnormal and insoluble aggregated proteins. Tremendous effort has been made to introduce therapeutic interventions to target tau and Aβ aggregates in AD patients. However, the processes involved in the accumulation of these proteins and the factors involved in the toxicity caused by them should be given special attention. Although apoptosis is known to be a consequence of deposition of aggregated proteins, recent evidence suggests the role of apoptosis-related factors in other aspects of AD pathophysiology. It is clearly understood that alterations in the activity of caspases as the main players in apoptosis play a crucial role in AD progression. Different caspases are involved in the processing of AβPP to generate Aβ plaques. Also, these proteases induce synaptic loss through cleavage of other intermediate substances in amyloidogenic pathway. In addition to Aβ accumulation, caspases play a central role in tauopathies as well as altered autophagy process and neuroinflammation as other main pathologic changes in AD. One of the causes of the change in the caspases activity in AD can be related to alteration is their regulators, especially miRs which should be given more attention. This evidence suggests the importance of caspases as therapeutic targets in AD. Indeed, a variety of caspase modulators have been observed to slow AD progression and improve cognitive impairment, at least in different AD models. However, the beneficial effects of caspase modulators in AD patients have not been investigated yet. On the other hand, although the effect of FDA-approved drugs of AD on caspases has been well studied, their combination with other compounds may have some benefits and can be investigated in clinical trials. Additionally, there are numerous caspase modulators with low side effects in pre-clinical studies which can be used in clinical studies to investigate their effect on the progression of AD.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0873r1).