
Abstract
Background:
Alzheimer’s disease (AD) is characterized by cognitive dysfunction and memory loss that is accompanied by pathological changes to white matter. Some clinical and animal research revealed that AD combined with chronic cerebral hypoperfusion (CCH) exacerbates AD progression by inducing blood-brain barrier dysfunction and fibrinogen deposition. Rivaroxaban, an anticoagulant, has been shown to reduce the rates of dementia in atrial fibrillation patients, but its effects on white matter and the underlying mechanisms are unclear.
Objective:
The main purpose of this study was to explore the therapeutic effect of rivaroxaban on the white matter of AD+CCH mice.
Methods:
In this study, the therapeutic effects of rivaroxaban on white matter in a mouse AD+CCH model were investigated to explore the potential mechanisms involving fibrinogen deposition, inflammation, and oxidative stress on remyelination in white matter.
Results:
The results indicate that rivaroxaban significantly attenuated fibrinogen deposition, fibrinogen-related microglia activation, oxidative stress, and enhanced demyelination in AD+CCH mice, leading to improved white matter integrity, reduced axonal damage, and restored myelin loss.
Conclusions:
These findings suggest that long-term administration of rivaroxaban might reduce the risk of dementia.
Keywords
INTRODUCTION
Recent studies suggest that pathological changes in white matter contribute to an increase in the risk and progression of cognitive dysfunction and memory loss in Alzheimer’s disease (AD) [1, 2]. Chronic cerebral hypoperfusion (CCH) induces a reduction in cerebral blood flow and hypoperfusion-associated blood-brain barrier (BBB) dysfunction, which are considered common features of AD, and is known to exacerbate disease progression by contributing to white matter damage [3 –5]. Some findings suggested that the breakdown of paranodal septate-like junctions relate to nodal structure changed when white matter was damaged [6 –8]. Fibrinogen, a coagulation factor that contains multiple binding sites for receptors, exists mainly in blood as a soluble protein, but can form fibrin clots when there is vessel damage and BBB leakage [9]. However, recent studies revealed that abnormal fibrinogen deposition in the cerebral nervous system (CNS) plays a considerable role in neurological diseases associated with BBB leakage, such as AD, stroke, brain trauma, and multiple sclerosis [10 –13]. The deposition of fibrinogen and its degradation products in the parenchyma have the ability to react with amyloid-β (Aβ) to form Aβ-fibrin clots, which are resistant to fibrinolysis, promote microglial activation that leads to enhanced inflammation and oxidative stress responses, and suppress remyelination and white matter damage [14 –19]. The AD+CCH model established in our previous studies displayed severe cognitive conflict accompanied by white matter damage and Aβ deposition [20, 21], and was able to mimic AD combined with cerebrovascular disease in this study. A recent study on the incidence of dementia in patients with atrial fibrillation undergoing anticoagulant therapy indicated that among 60,178 patients treated with warfarin, the incidence of dementia was 27.3 (per 1000 person-years) over a 1.4-year follow-up period. An equal number of patients treated with rivaroxaban showed an incidence of 22.2 (per 1000 person-years) over a 1.2-year follow-up period [22]. Other clinical reports also offered support of lower rates of dementia among atrial fibrillation patients after initiating rivaroxaban treatment relative to warfarin treatment [23, 24]. In several recent studies, rivaroxaban, but not warfarin, displayed anti-inflammatory properties by improving protease-activated receptor (PAR)-1 and -2 in vascular disease while reducing intracerebral hemorrhage [25 –29]. However, there are still only limited studies dedicated to the treatment effects of anti-coagulants on dementia.
In our previous study, we found that rivaroxaban treatment attenuated cerebral hemorrhage, BBB leakage, and cognitive dysfunction in the cortex and hippocampus of an AD+CCH mouse model, although the effect of rivaroxaban on white matter and the possible underlying mechanism were not entirely clear [25]. The main purpose of this study was to explore the therapeutic effect of rivaroxaban on the white matter of AD+CCH mice. To investigate the possible mechanism, focus was placed on changes in fibrinogen deposition, as well as the effects of fibrinogen-related inflammation and oxidative stress on oligodendrocytes and oligodendrocyte precursor cells in white matter.
MATERIALS AND METHODS
Animals
All procedures were conducted in accordance with the Animal Committee of the Graduate School of Medicine and Dentistry of Okayama University under the authority of project license number OKU-2018-364 and ARRIVE guidelines (https://www.nc3rs.org.uk/arrive-guidelines) as well as the Okayama University guidelines on the Care and Use of Laboratory Animals. The present study is a part of a larger project focusing on the effect of rivaroxaban in the AD+CCH model [25, 28]. To establish the AD+CCH model, we used APP23 transgenic AD mice that overexpress human Swedish mutant amyloid precursor protein, which presents both a parenchymal senile plaque and cerebral amyloid angiopathy (CAA) [30 –32]. All mice had a C57BL/6J genetic background. Employing an effect size from previous experiments of 1.11, α= 0.05 and 90% power, a sample size of 4 mice per group was needed. In total, 10 WT mice (5 male and 5 female) and 44 APP23 mice (30 male and 14 female) were used in this study. The exclusion criteria for this study were as follows: mice that died as a result of procedural problems during CCH surgery or after surgery (n = 26); mice that failed to display a decrease in CBF after CCH surgery (n = 2). No mice died during CCH surgery and the mortality rate after CCH surgery until sacrifice in this study was 59%. Buprenorphine (0.05 mg/kg, 0.015 mg/mL) was intramuscularly injected into mice after operation to relieve pain. The final number of mice in the four experimental groups that were sacrificed were: Wild type (WT) plus sham surgery group (WT, n = 10), APP23 mice plus CCH surgery group (APP+CCH, n = 5), APP23 mice plus CCH surgery plus warfarin treatment group (APP+CCH+W, n = 5), and APP23 mice plus CCH surgery plus rivaroxaban treatment group (APP+CCH+R, n = 6). All mice were housed in a 12-h day/night cycle with controlled temperature and ad libitum access to liquid gel (MediDrop® Sucralose, ClearH2O, Westbrook, ME, USA) and a standard laboratory diet (MF; Oriental Yeast, Tokyo, Japan), the maximum number of mice in a cage was 5.
Chronic cerebral hypoperfusion model
Chronic cerebral hypoperfusion was performed using ameroid constrictors (ACs) with an inner diameter of 0.75 mm (Research Instruments NW, Lebanon, OR, USA) to achieve a gradual and progressive decrease in cerebral blood flow (CBF) at 4 months (M) of age, as in our previous studies [20, 33]. Briefly, mice were anesthetized with a mixture of nitrous oxide/oxygen/isoflurane (69% :30% :1.5%) via an inhalation mask with a constant temperature (37°C). Both common carotid arteries were exposed through a midline incision and ACs were implanted into vessels. Mice in the sham group underwent the same surgical procedure, but without ACs. CBF was measured with a laser-doppler flowmeter (FLO-C1, Omegawave, Tokyo, Japan) at 1, 3, 7, 14, and 28 days after surgery.
Warfarin and rivaroxaban administration
Warfarin and rivaroxaban powder were mixed into a liquid gel and provided to mice orally through bottled drinking gel. Administration of the warfarin and rivaroxaban mixture started 15 days after surgery, and lasted until sacrifice, at 10 months. The optimal dosages of warfarin (0.2 mg/kg/day) and rivaroxaban (60 mg/kg/day) were determined in our previous study [25].
Tissue preparation
Mice of all groups were deeply anesthetized by intraperitoneal injection of pentobarbital (40 mg/kg), then transcardially perfused with ice-cold phosphate-buffered saline (PBS, pH 7.4), followed by 4% ice-cold paraformaldehyde (PFA) in PBS. Brain tissues were transferred into PBS containing 10, 20 and 30% (w/v) sucrose for 24 h at 4°C after post-fixation in 4% PFA overnight, then cut into 20μm thick coronal sections with a cryostat at –22°C.
Immunohistochemistry
To determine morphological and pathological changes in white matter, damage to the corpus callosum (CC) was determined with luxol fast blue (LFB) staining using the LFB Stain Kit (LBC-1; ScyTek Laboratories, Inc.; Logan, UT, USA). For LFB staining, sections were immersed in distilled water then incubated in Luxol Fast Blue solution for 24 h at room temperature. After rinsing thoroughly in distilled water, sections were dipped twice in lithium carbonate solution (0.05%) 20 s each dip. Sections were then soaked in 70% alcohol for 10 min and dehydrated in three changes of absolute alcohol before mounting on slides.
For single immunohistochemistry, antigen retrieval was performed using 10 mM citric buffer (pH 6.0) in a microwave at 500 W for 2 min. After cooling, brain sections were immersed in 0.3% hydrogen peroxide/PBS for 30 min to block the intrinsic activity of peroxidases, then incubated in 5% bovine serum albumin (BSA) in PBS for 1 h. The following primary antibodies were incubated with brain sections overnight at 4°C: rabbit anti-myelin basic protein (MBP) antibody (1:500, ab40390; Abcam, Cambridge, UK), mouse anti-myelin-associated glycoprotein (MAG) antibody (1:200, sc-166849; Santa Cruz Biotechnology, San Jose, CA, USA), rabbit anti-fibrinogen antibody (1:100, ab34269; Abcam), mouse anti-4-HNE antibody (1:50, MHN-020P; JaICA, Shizuoka, Japan), mouse anti-8-OHdG (1:50, MOG-020P; JaICA), rabbit anti-APC antibody (1:200, ab72040; Abcam). After incubation with primary antibodies, sections were washed once in PBS and incubated at room temperature for 2 h with biotinylated secondary antibodies, anti-mouse antibody (1:500, PK-4002, Vector Laboratories, Newark, NJ, USA) and anti-rabbit antibody (1:500, PK-4001, Vector Laboratories), against a host of primary antibodies, including MBP, MAG, fibrinogen, 4-HNE, 8-OHdG, and APC. Signal amplification was performed using the Vectastain Elite ABC Kit (PK-6100; Vector Laboratories) for 30 min and visualized with 3,3′-diaminobenzidine (045-22833; Fujifilm). Negative control sections were stained in the same manner but without any primary antibody. Slides were digitized under a light microscope (Olympus BX-51, Tokyo, Japan).
For double immunofluorescence staining, antigen was retrieved in 10 mM citric buffer (pH 6.0) after microwaving for 2 min. After cooling, brain sections were incubated in 5% BSA in PBS for 1 h. The following primary antibodies were incubated with brain sections overnight at 4°C: mouse anti-Caspr antibody, clone K65/35 (1:100, MABN9; Merck Millipore, Burlington, MA, USA), rabbit anti-Nav1.6 antibody (1:200, AB5580; Merck Millipore), rabbit anti-MBP antibody (1:500, ab40390; Abcam), mouse anti-SMI32 antibody (1:100, 801701; Biolegend, San Diego, CA, USA), rabbit anti-fibrinogen antibody (1:100, ab34269; Abcam), rat anti-CD11b/ITGAM (M1/70) (CD11b) antibody (1:50, 46512 s; Cell Signaling Technology, Massachusetts, MA, USA), rabbit anti-Ki67 antibody (1:500, ab15580; Abcam), or goat anti-PDGFRα antibody (1:100, AF1062; R&D Systems, Minneapolis, MN, USA). After incubation, sections were washed once in PBS and incubated (room temperature for 2 h) with secondary antibodies, Alexa Fluortrademark 555 donkey-anti-mouse (H + L) antibody (1:500, A31570; Invitrogen, Carlsbad, CA, USA), Alexa Fluortrademark 488 donkey-anti-rabbit (H + L) antibody (1:500, A21206; Invitrogen), Alexa Fluortrademark 594 donkey-anti-rabbit (H + L) antibody (1:500, A21207; Invitrogen), Alexa Fluortrademark 488 donkey-anti-rat (H + L) antibody (1:500, A21208; Invitrogen), against a host of primary antibodies, including Caspr, Nav1.6, SMI32, fibrinogen, CD11b, Ki67, and PDGFRα. After washing with PBS, sections were treated with a TrueBlack lipofuscin autofluorescence quencher (Biotium, San Francisco, CA, USA) to block lipofuscin autofluorescence. Negative control sections were stained in the same manner, but without any primary antibody. Slides were mounted with Vectashield mounting medium containing DAPI (Vector Laboratories) and digitized under a confocal microscope (LSM-780; Zeiss, Jena, Germany).
Semiquantitative analysis
For the semiquantitative analysis of LFB and single immunohistochemistry, background subtraction was performed by applying a constant threshold value, and pixel intensity was measured from three sections and four randomly selected regions from each mouse brain. For the semiquantitative analysis of fibrinogen/CD11b and Ki67/PDGFRα staining, the number of positive cells in the CC was counted. The ratio of SMI32 and MBP pixel intensities was calculated to assess demyelination and axon damage.
Statistical analysis
Three sections per mouse were used from all frozen sections for immunohistochemistry, and four randomly selected sites were measured per section. The length of the Nav1.6 channel in the CC, as well as the gap length between Caspr signals next to the Nav1.6 signal, were measured and analyzed by immunofluorescence staining. All immunostaining data were analyzed by image processing software (Image J, Bethesda, MD, USA). Statistical analysis was performed in GraphPad Prism (version 8.3, GraphPad Software Inc., San Diego, CA, USA). All results, which were normally distributed, were expressed as the mean±standard error of the mean (SEM). Statistical comparisons of normally distributed data involved a one-way analysis of variance (ANOVA) followed by the Tukey–Kramer test. Statistical significance was assessed at p < 0.05. Analysis was performed blindly with the investigator unaware of experimental groups.
RESULTS
CCH surgery reduced CBF in AD mice
CCH surgery reduced CBF in APP+CCH, APP+CCH+W, and APP+CCH+R groups to 65% of the baseline. At 15 days after surgery, warfarin and rivaroxaban were administered to APP+CCH+W and APP+CCH+R groups. There were no significant differences in CBF between these three groups at 28 days [25].
Rivaroxaban improved white matter integrity in the corpus callosum
Both LFB staining (Fig. 1a), as well as immunostaining of MBP (Fig. 1b) and MAG (Fig. 1c), were used to evaluate the severity of myelin injury in the CC. Myelin fibers appeared blue in LFB staining. MBP and MAG are proteins known to play important roles in the myelination of nerves. Demyelination was significantly more severe in both APP+CCH and APP+CCH+W groups than in the WT group. However, rivaroxaban treatment significantly rescued the loss of myelin in the CC (Fig. 1d, # p < 0.05 versus APP+CCH; $ p < 0.05 versus APP+CCH+W, Tukey’s multiple comparison test). The results of MBP and MAG staining also indicate that the APP+CCH+R group showed significantly less myelin damage than the APP+CCH+W group (Fig. 1b, c, e, and f, $ p < 0.05 and $$ p < 0.01 versus APP+CCH+W).
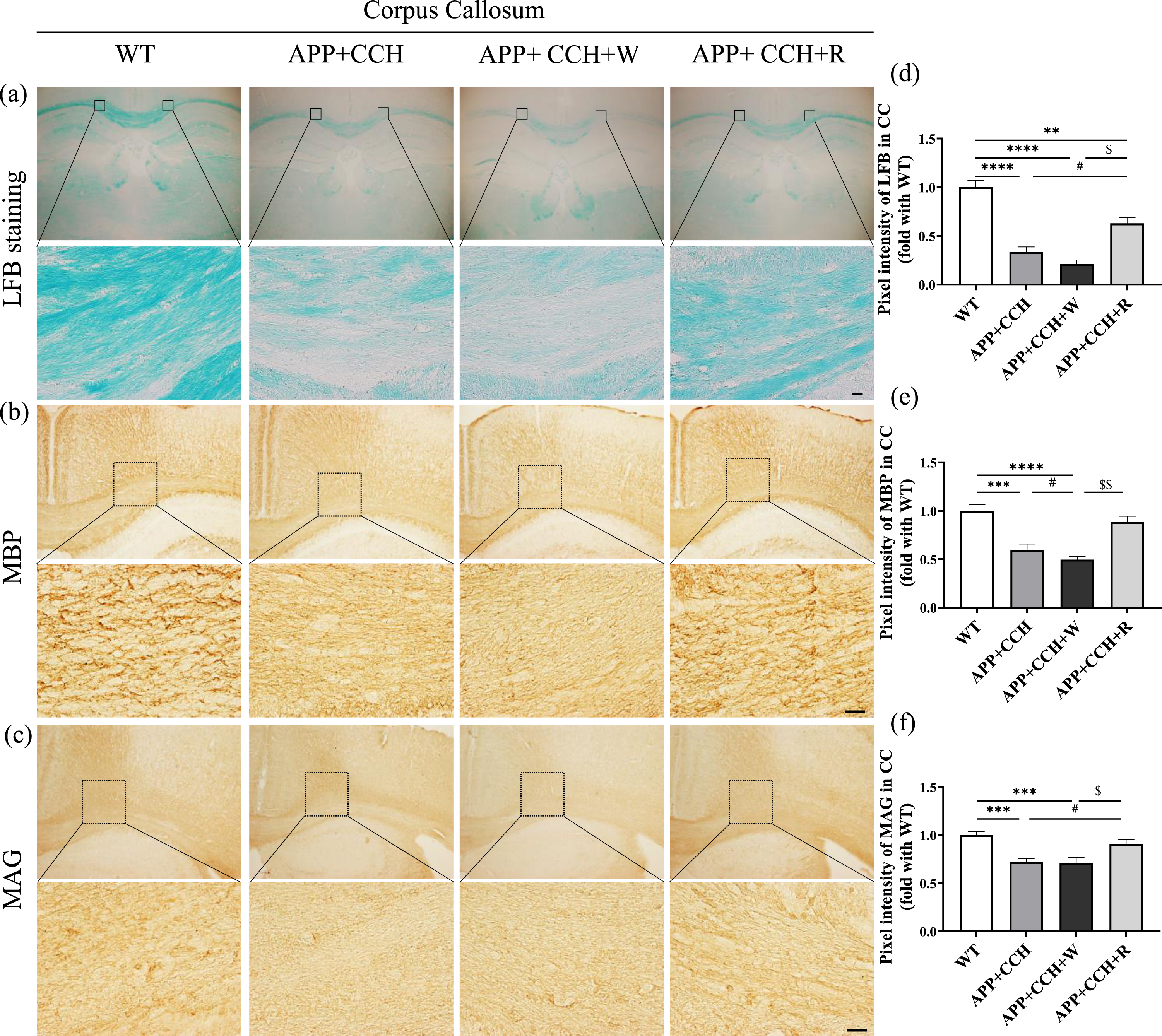
(a–c) Myelin fiber loss and the expression level of myelin-related protein in the corpus callosum (CC) revealed by LFB staining and single immunohistochemistry of myelin basic protein (MBP) and myelin-associated glycoprotein (MAG). Scale bar = 20μm. (d-f) Quantitative analysis of pixel intensities of LFB, MBP, and MAG staining. Note the significant recovery of LFB and MAG pixel intensities in the APP+CCH+R group compared with the APP+CCH group (**p < 0.01, ***p < 0.001, and ****p < 0.0001 versus WT; # p < 0.05 versus APP+CCH; $ p < 0.05 and $$ p < 0.01 versus APP+CCH+W).
Double staining of SMI32 and MBP was used to assess the damage and demyelination of axons in the CC (Fig. 2a, b). SMI32 is a neurofilament H non-phosphorylated type of marker that can be observed in thick damaged axons [34]. The results of SMI32 and MBP staining suggest that severe injury and demyelination occurred to the axons of the APP+CCH and APP+CCH+W groups in the CC and striatum (ST) while axonal injury and demyelination were less severe in the APP+CCH+R group (Fig. 2c and d, *p < 0.05, ***p < 0.001, and ****p < 0.0001 versus WT; $$ p < 0.01 and $$$$ p < 0.0001 versus APP+CCH+W).
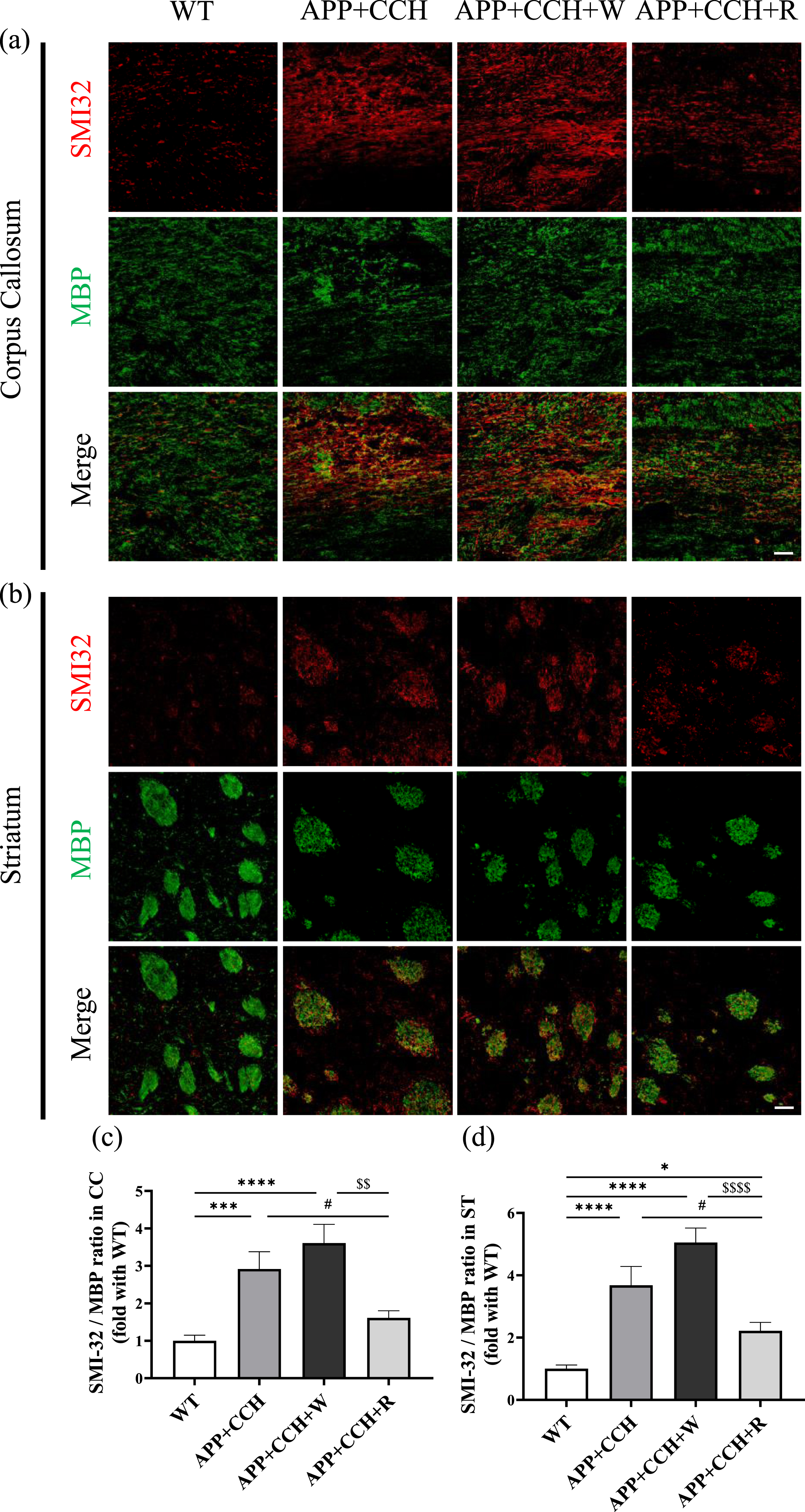
(a, b) Demyelinated and damaged axons in the corpus callosum (CC) and striatum (ST) revealed by double immunostaining of MBP and SMI32. Scale bar = 20μm. (c, d) Quantitative analysis of the ratio of pixel intensities of SMI32 and MBP staining. Note the significant decrease in the ratio of pixel intensities of SMI32 and MBP staining in the APP+CCH+R group compared to the APP+CCH group (*p < 0.05, ***p < 0.001, and ****p < 0.0001 versus WT; # p < 0.05 versus APP+CCH; $$ p < 0.01 and $$$$ p < 0.0001 versus APP+CCH+W).
Rivaroxaban suppressed extension of the Nav1.6 channel in the corpus callosum
To evaluate damage to paranodal integrity in white matter, double immunofluorescence staining of Nav1.6 and Caspr were performed to reveal the structural changes to Ranvier nodes and the paranode in the CC. The Nav1.6 channel cluster is one molecular component of the nodes of Ranvier, which participate in the regulation of nerve conduction velocity. Caspr is a membrane protein that is mainly is observed in CNS myelinated nerve fibers within paranodal axoglial junctions. The Nav1.6 channel cluster is located in the nodes of Ranvier and is flanked by Caspr, a paranode membrane protein. The Nav1.6 channel cluster progressively extended into both sides of the paranode in both the APP+CCH and APP+CCH+W groups (Fig. 3a). However, the APP+CCH+R group showed less extension than the APP+CCH+W group (Fig. 3b, *p < 0.05 and ***p < 0.001 versus WT; $ p < 0.05 versus APP+CCH+W). There was a significant difference in Caspr gap length between the WT and APP+CCH+W groups (Fig. 3c, **p < 0.01 versus WT). There were no significant differences in the number of nodes between all groups (Fig. 3d).
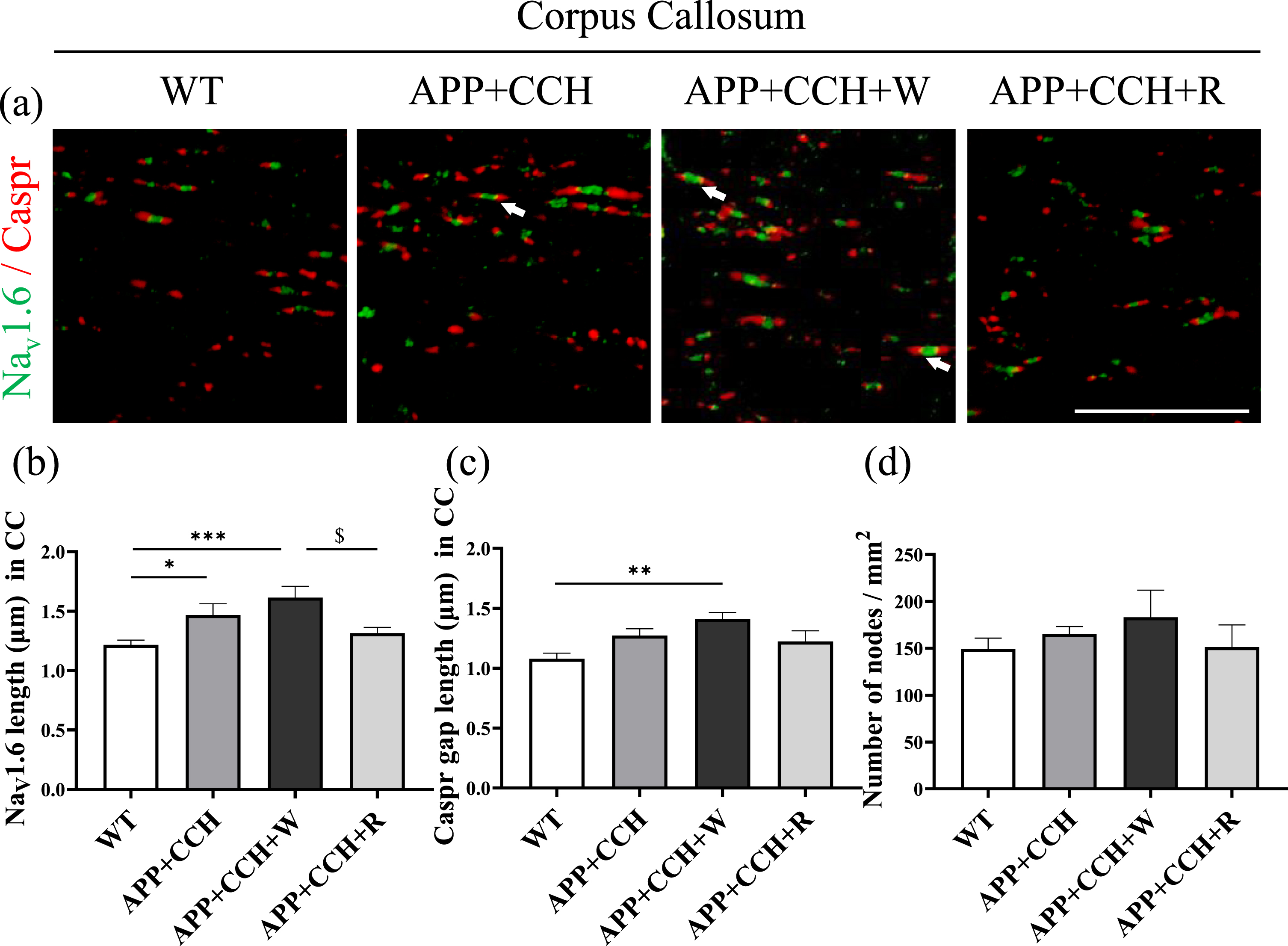
(a) Paranodal integrity damage in the corpus callosum (CC) revealed by double immunostaining of Nav1.6 and Caspr. Scale bar = 20μm. Arrowheads represent an extension of the Nav1.6 channel beyond the primary nodal region. (b-d) Quantitative analysis of Nav1.6 length, Caspr gap length, and number of nodes of Nav1.6 and Caspr staining. Note the significant decrease of Nav1.6 length in the APP+CCH+R group compared to the APP+CCH+W group (*p < 0.05, **p < 0.01, and ***p < 0.001 versus WT; $ p < 0.05 versus APP+CCH+W).
Rivaroxaban enhanced the proliferation of oligodendrocyte precursor cells in the corpus callosum
The analysis of pixel intensity after staining with an oligodendrocyte marker (APC) revealed a considerable loss of oligodendrocytes in the APP+CCH and APP+CCH+W groups. However, rivaroxaban treatment attenuated this loss (Fig. 4c, *p < 0.05 and ***p < 0.001 versus WT; $ p < 0.05 versus APP+CCH+W). PDGFRα is a receptor located on the surface of cell, specifically expressed by oligodendrocyte progenitor cell (OPC) in the CNS and was used as an OPC marker. Compared with the WT group, the number of PDGFRα-positive cells decreased significantly in the APP+CCH and APP+CCH+W groups (Fig. 4d, **p < 0.01 versus WT). In addition, rivaroxaban treatment dramatically increased the number of PDGFRα/Ki67 double-positive cells in the APP+CCH+R group compared with the APP+CCH+W and APP+CCH groups (Fig. 4e, # p < 0.05 versus APP+CCH; $$ p < 0.01 versus APP+CCH+W).
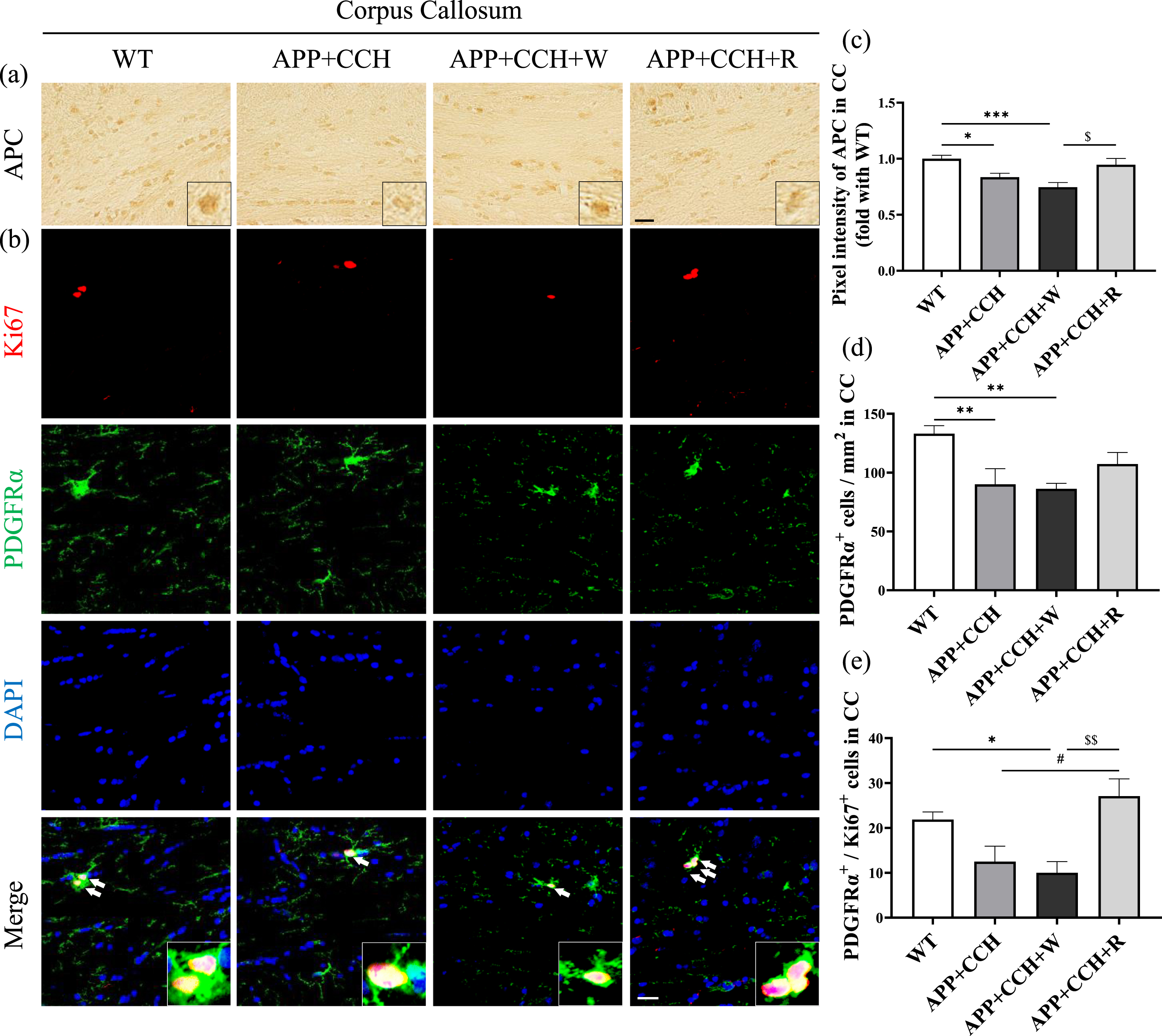
(a, b) Expression level of oligodendrocytes and the proliferation level of oligodendrocyte precursor cells (OPC) in the corpus callosum (CC) were revealed by single immunohistochemistry of an oligodendrocyte cell marker (APC), as well as double staining of an OPC marker (PDGFRα) and a cell proliferation marker (Ki67). Scale bar = 20μm. Arrowheads represent PDGFRα/Ki67 double-positive cells. (c–e) Quantitative analysis of pixel intensities of APC as well as PDGFRα-positive and PDGFRα/Ki67 double-positive cell number. Note the significant increase in the number of PDGFRα/Ki67 double-positive cells in the APP+CCH+R group compared to the APP+CCH group (*p < 0.05, **p < 0.01, and ***p < 0.001 versus WT; # p < 0.05 versus APP+CCH; $ p < 0.05 and $$ p < 0.01 versus APP+CCH+W).
Rivaroxaban attenuated fibrinogen deposition induced by oxidative stress in the corpus callosum
Fibrinogen staining revealed that fibrinogen deposition increased in the APP+CCH and APP+CCH+W groups, but rivaroxaban treatment significantly rescued fibrinogen deposition in the APP+CCH+R group (Fig. 5d, **p < 0.01 and ***p < 0.001 versus WT; $ p < 0.05 versus APP+CCH+W). Staining with a lipid peroxidation marker (4-HNE) and a DNA oxidation marker (8-OHdG) revealed a considerable increase in their expression in the APP+CCH and APP+CCH+W groups, but rivaroxaban treatment dramatically reversed the increase in 8-OHdG expression (Fig. 5e and f, *p < 0.05, ***p < 0.001, and ****p < 0.0001 versus WT; $$$$p < 0.0001 versus APP+CCH+W).
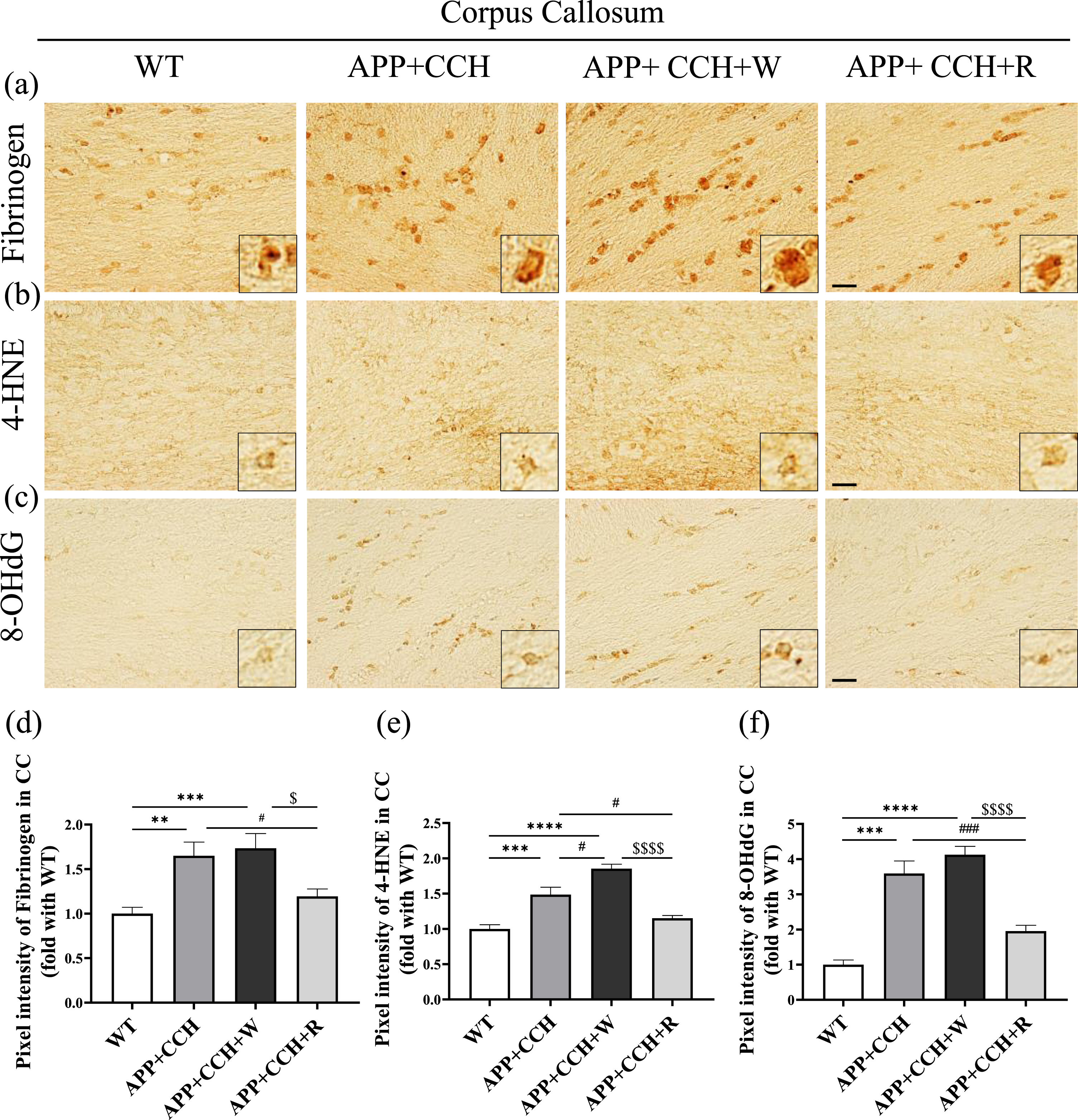
(a) Expression level of fibrinogen deposition and oxidative stress in the corpus callosum (CC) evaluated by single immunohistochemistry. (b, c) The level of oxidative stress was assessed by a lipid peroxidation marker (4-HNE) and a nucleic acid peroxidation marker (8-OHdG). (d–f) Quantitative analysis of pixel intensities of fibrinogen, 4-HNE, and 8-OHdG. Note the significant decrease in pixel intensities of fibrinogen, 4-HNE, and 8-OHdG in the APP+CCH+R group compared with the APP+CCH group (**p < 0.01, ***p < 0.001, and ****p < 0.0001 versus WT; # p < 0.05 and # # # p < 0.001 versus APP+CCH; $ p < 0.05 and $$$$ p < 0.0001 versus APP+CCH+W).
CD11b-positive microglia show mediation effects on several immune processes such as phagocytosis, cell-mediated cytotoxicity, chemotaxis, companied with morphological changes and express more inflammatory factors [35]. Activation of CD11b-positive microglia was detected in the APP+CCH, APP+CCH+R, and APP+CCH+W groups, but the number of CD11b-positive cells in the APP+CCH+R group was significantly less than in the APP+CCH+W group (Fig. 6b, **p < 0.01, ***p < 0.001, and ****p < 0.0001 versus WT; $$ p < 0.01 versus APP+CCH+W). Activation of fibrinogen-related CD11b-positive microglia increased significantly in the APP+CCH group and was significantly aggravated in the APP+CCH+W group. In contrast, the APP+CCH+R group showed a significant decrease in cell number relative to the APP+CCH+W group (Fig. 6c, ***p < 0.001, and ****p < 0.0001 veresus WT; # p < 0.05 versus APP+CCH; $$$ p < 0.001 versus APP+CCH+W).
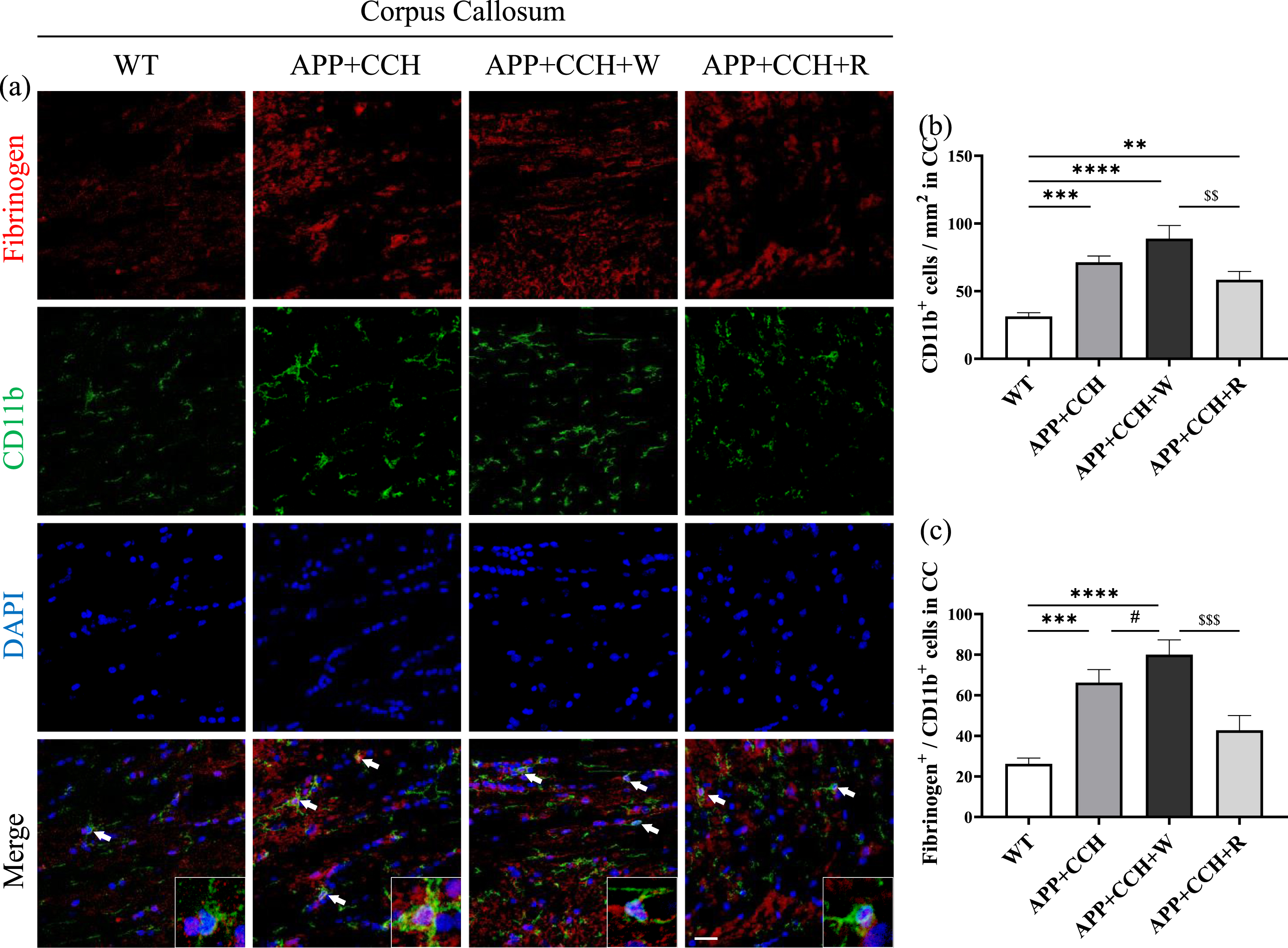
(a) Activation of CD11b-positive microglia and fibrinogen-related CD11b-positive microglia in the corpus callosum (CC). Scale bar = 20μm. (b, c) Quantitative analysis of the number of CD11b-positive cells and CD11b/fibrinogen double-positive cells. Note the significant decrease in the number of CD11b/fibrinogen double-positive cells in the APP+CCH+R group compared to the APP+CCH+W group (**p < 0.01, ***p < 0.001, and ****p < 0.0001 versus WT; # p < 0.05 versus APP+CCH; $$ p < 0.01 and $$$ p < 0.001 versus APP+CCH+W).
DISCUSSION
The present study reports, for the first time, differences in treatment effects on white matter following long-term administration of rivaroxaban and warfarin. This experiment also explored the potential underlying mechanisms in a mouse model of AD combined with cerebral hypoperfusion, i.e., AD+CCH. These results demonstrate that rivaroxaban dramatically alleviated fibrinogen deposition, fibrinogen-related microglia activation, oxidative stress, and enhanced oligodendrocyte precursor cell proliferation in the white matter of AD+CCH mice (Figs. 4–6). Male and female mice number in WT, APP+CCH, APP+CCH+W, and APP+CCH+R groups were: 5:5; 4:1; 4:1; 4:2, separately. Female mice showed higher mortality in APP23 mice probably because female could have more aggressive AD pathology. These effects might contribute to the protection of myelin and reduce axon damage and abnormal changes to paranode structure compared to warfarin treatment (Figs. 1–3).
Previous clinical research reported a relationship between fibrinogen and AD pathology, revealing higher levels of fibrinogen and its degraded production in both the parenchyma and serum of AD patients [36, 37]. Multiple studies have suggested that fibrinogen in the CNS may contribute to the pathology of AD in animal models through various neuropathological mechanisms, such as the induction of microglial activation, or inhibition of OPC differentiation, causing axonal damage and demyelination, and interacting with Aβ, suggesting that the pathological effects of fibrinogen on AD are associated with damage to white matter [17 , 38–40]. In the present study, an AD+CCH mice model, which exhibits significant injury to white matter, was used to investigate the influence of rivaroxaban and warfarin treatments on white matter [20 , 41]. Previous studies suggested that a higher proportion of non-phosphorylated SMI32+ neurofilaments and MBP could be found in demyelinated and damaged axons [42]. Our findings indicate that rivaroxaban treatment, but not warfarin, protected the integrity of white matter in the AD+CCH mouse model, reducing the loss of myelin and restoring the expression of key myelin-related proteins, especially MAG. Furthermore, rivaroxaban treatment improved damage to myelinated axons in the CC and ST of AD+CCH mice (Figs. 1 and 2). Structural changes in the nodes of Ranvier and paranode were primarily observed in the APP+CCH and APP+CCH+W groups, indicating the breakdown of paranodal septate-like junctions [20]. Rivaroxaban treatment resulted in better outcomes than warfarin by preventing the extension of Nav1.6. On the other hand, there was no significant difference in Caspr gap length, probably because the APP+CCH mice were too young to show a considerable change after 10 months (Fig. 3).
Next, we sought to appreciate the possible mechanism by which rivaroxaban attenuated damage to white matter, including axonal damage and demyelination. As expected, an increase in fibrinogen accumulation was observed in the white matter of APP+CCH mice following BBB injury (Fig. 5) [43, 44]. Additionally, enhanced oxidative stress and fibrinogen-induced microglia activation, as well as a decrease in OPC number, were observed in APP+CCH mice (Figs. 4–6). Fibrinogen was reportedly able to induce microglial activation by binding to CD11b, indicating that increasing fibrinogen deposition may induce microglial activation and aggravate oxidative stress and inflammation in CC [18 , 45–47], as was also observed in the present study. Moreover, since BBB leakage occurred in the AD+CCH model, the increase in CD11b-positive cells might also suggest the infiltration of peripheral leukocytes, which may cause autoimmune responses and contribute to white matter damage in the CNS [18]. Previous studies also suggested that fibrinogen deposition, oxidative stress, and inflammation inhibited OPC proliferation in neurodegenerative disease, and that this could cause remyelination failure and more serious myelin loss, leading to white matter damage [17 , 49]. Fibrinogen also directly inhibited OPC proliferation via the BMP pathway [17, 39]. As a result, a reduced number of oligodendrocytes and OPC led to impaired remyelination and aggravated white matter damage. Our previous findings suggested that rivaroxaban improved BBB leakage via an inhibitory effect of rivaroxaban on protease-activated receptor (PAR)-1 and PAR-2, causing less fibrinogen to enter the brain and be deposited there. However, warfarin treatment induced more cerebral microbleeds and greater fibrinogen leakage [25, 28]. The activation of PAR-1 and PAR-2 could induce oxidative stress and inflammation by activating microglia through extracellular signal-regulated kinase pathways. Activated PAR-1 may inhibit neurons from developing synaptic plasticity [50 –54]. Moreover, a recent study suggested that amyloid plagues might not be the prime cause of inflammation and cognitive conflicts in AD. Rather, microglia may play an important role before amyloid plaques form, and are highly associated with their formation [55], lending support to our findings. Therefore, we suggest that an appropriate long-term application of rivaroxaban in the elderly population may potentially reduce the risk of dementia caused by fibrinogen-induced white matter damage.
Recently, two new anti-amyloid immunotherapy treatments, Aducanumab and Lecanemab, which were approved by the US Food and Drug Administration for treating AD, are promising candidates since they significantly reduced cerebral amyloid deposits [56 –59]. However, these treatments carry a significant risk of amyloid-related imaging abnormality (ARIA). ARIA has two subtypes, ARIA-E type, characterized by vasogenic edema or sulcal effusion, and ARIA-H type, characterized by hemosiderin deposits involving microhemorrhages and superficial siderosis, identified by magnetic resonance imaging [60, 61]. In patients with AD requiring anticoagulation therapy, including those undergoing anti-amyloid therapy, rivaroxaban may be a suitable candidate for administration considering its protective effect against BBB damage and ability to attenuate amyloid pathology and microhemorrhages [25].
There are limitations to this experiment, which found a therapeutic effect of rivaroxaban in the AD+CCH model. The treatment effects of rivaroxaban in APP23 or WT+CCH mice remain unknown. Moreover, apixaban showed a decreased risk of major ischemic or hemorrhagic events in AF patients compared with rivaroxaban [62]. The mechanism underlying the differences between these two anticoagulants is still unclear, so additional studies are needed to reveal it in detail.
In conclusion, this study demonstrated a protective effect of rivaroxaban on white matter integrity, being able to rescue remyelination failure in a novel AD+CAA mice model. Fibrinogen may play an essential role in the aggravation of microglial activation and following oxidative stress and inflammation. This aggravation was partly rescued by the administration of rivaroxaban but not warfarin. Therefore, these findings suggest that rivaroxaban can not only be used to reduce the risk of dementia, but also serve as a potential candidate for combination therapy with anti-amyloid immunotherapy.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This study was partly supported by a Grant-in-Aid for Scientific Research (C) 20K09370, 20K12044, Challenging Research 21K19572, Young Research 20K19666, 21K15190, and by Grants-in-Aid from the Research Committees (Toba K, and Tsuji S) from the Japan Agency for Medical Research and Development.
CONFLICT OF INTEREST
Toru Yamashita is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.