
Abstract
Background:
Apolipoproteins and contactin 5 are proteins associated with Alzheimer’s disease (AD) pathophysiology. Apolipoproteins act on transport and clearance of cholesterol and phospholipids during synaptic turnover and terminal proliferation. Contactin 5 is a neuronal membrane protein involved in key processes of neurodevelopment.
Objective:
To investigate the interactions between contactin 5 and apolipoproteins in AD, and the role of these proteins in response to neuronal damage.
Methods:
Apolipoproteins (measured by Luminex), contactin 5 (measured by Olink’s proximity extension assay), and cholesterol (measured by liquid chromatography mass spectrometry) were assessed in the cerebrospinal fluid (CSF) and plasma of cognitively unimpaired participants (n = 93). Gene expression was measured using polymerase chain reaction in the frontal cortex of autopsied-confirmed AD (n = 57) and control subjects (n = 31) and in the hippocampi of mice following entorhinal cortex lesions.
Results:
Contactin 5 positively correlated with apolipoproteins B (p = 5.4×10–8), D (p = 1.86×10–4), E (p = 2.92×10–9), J (p = 2.65×10–9), and with cholesterol (p = 0.0096) in the CSF, and with cholesterol (p = 0.02), HDL (p = 0.0143), and LDL (p = 0.0121) in the plasma. Negative correlations were seen between CNTN5, APOB (p = 0.034) and APOE (p = 0.015) mRNA levels in the brains of control subjects. In the mouse model, apoe and apoj gene expression increased during the reinnervation phase (p < 0.05), while apob (p = 0.023) and apod (p = 0.006) increased in the deafferentation stage.
Conclusions:
Extensive interactions were observed between contactin 5 and apolipoproteins and cholesterol, possibly due to neuronal damage. The alterations in gene expression of apolipoproteins suggest a role in axonal, terminal, and synaptic remodeling in response to entorhinal cortex damage.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of degenerative dementia affecting over 33 million people worldwide [1]. AD is defined by the presence of amyloid-β (Aβ) plaques and pathologic tau [2] that leads to cognitive decline. While the majority of research has focused on the pathological processes that lead to Aβ and tau deposition and neuronal death, less attention is given to the changes that occur in response to this pathological process in an attempt to promote compensatory changes and reinnervation.
Current literature suggests that apolipoproteins play a central role in the compensatory response to neuronal damage, possibly due to their role in the transport and clearance of cholesterol and phospholipids during synaptic turnover and terminal proliferation [3]. The main apolipoproteins involved in cholesterol transport in the central nervous system are apolipoproteins E (APOE), B (APOB), D (APOD), and J (APOJ). All of them have been previously implicated in different aspects of AD pathophysiology [4–11]. The APOE ɛ4 allele is well known as the most important genetic risk factor for sporadic AD [12, 13], and the CLU (also called APOJ) gene is also currently associated with risk of AD [14, 15]. Rare genetic variants in the APOB gene have been associated with increased risk for early-onset familial AD [6]. Furthermore, apoB was found to be higher in the cerebrospinal fluid (CSF) of AD patients and was strongly related to alterations in tau and phospho-tau, as well as changes in synaptic proteins in asymptomatic subjects at risk of dementia [7]. Similarly, apoD was found to be increased in the CSF [5] and in the hippocampus of AD patients [5, 16].
Contactin 5 is another protein recently associated with sporadic AD risk and pathology. Contactin 5 is a neuronal membrane protein that plays crucial roles in the organization of axonal domains, axonal guidance, myelination, neuritogenesis, synaptogenesis and axo-glia interactions [17, 18]. Recently, it was shown that the rs1461684 G variant in the CNTN5 gene is associated with increased risk and a faster rate of progression throughout the AD spectrum [19]. Moreover, contactin 5 was found to increase progressively with age and to be associated with tau biomarkers and soluble synaptic proteins in the CSF [19].
In the present work, we sought to investigate the interplay between the soluble form of contactin 5 and apolipoproteins in AD. Investigating the connections between these two classes of proteins, which are related to synaptogenesis and synaptic reorganization, may provide us with a better understanding of how neurons respond to pathological harm and increase our understanding of AD pathophysiology.
METHODS
Study populations
Data were obtained from the Pre-symptomatic Evaluation of Experimental or Novel Treatments for Alzheimer’s Disease (PREVENT-AD) cohort and from the Quebec Founder Population (QFP) cohort. All procedures were approved by the McGill University Faculty of Medicine Institutional Review Board and complied with the ethical principles of the Declaration of Helsinki.
PREVENT-AD cohort
The PREVENT-AD cohort includes over 365 cognitively unimpaired subjects who have a first-degree relative with AD and therefore are at a higher risk of developing this dementia [20]. Participants are monitored annually with clinical and cognitive assessments, CSF and blood biomarkers and neuroimaging modalities (PET and MRI) [20]. Data used in the preparation of this article were obtained from the PREVENT-AD program https://douglas.research.mcgill.ca/stop-ad-centre), data release 6.0.
A complete listing of the PREVENT-AD Research Group can be found in the PREVENT-AD database: https://preventad.loris.ca/acknowledgements/acknowledgements.php?date=2023-04-01.
CSF
CSF was collected from a subset of participants of the PREVENT-AD cohort. In this study, samples from 93 subject, for whom we had all necessary measurements, were used. Lumbar punctures were performed in the morning following an overnight fast using a Sprotte 24-gauge atraumatic needle as described in Tremblay-Mercier et al (2021) [21]. CSF was centrifuged for 10 min at room temperature, cells and insoluble material were excluded, and aliquots were stored at –80°C.
Proteins
Apolipoproteins were measured using the apolipoprotein Luminex assay kit (10-plex magneto-fluorescent immunoassays, cat# 12003081, BioRad, USA) as per manufacturer instructions. Contactin 5 was measured using the neurology panel of Olink’s proximity extension assay as described before [19].
Cholesterol
CSF samples were homogenized briefly by sonication in butanol/methanol (3 : 1) and 4 M KOH in the presence of internal standards, followed by incubation at 37°C for 1.5 h. The second extraction was performed in heptane/ethyl acetate (3 : 1) and acetic acid (1%). The superior sterol-containing layer was vacuum dried and resuspended in 50μL of methanol (90%) and injected into RP-UPLC/MS. The chromatographic system was a Waters ACQUITY UPLC equipped with a Phenomenex Kinetics C18 column (2.1×150 mm 1.7μm). The Mass spectrometer is AB Sciex 6500 Qtrap. Data analysis software used is Analyst and Multiquant.
Blood
Blood samples were collected immediately after the lumbar puncture following an overnight fast. Blood was centrifuged for 10 min at 4°C, cells and insoluble material were saved, and plasma aliquots were stored at –80°C. Protein and cholesterol measurements were performed in plasma using the methods described above.
DNA extraction and genotyping
Genomic DNA was extracted from buffy coat using the Qiagen EZ1 DNA Kit. Genotype profiling of ApoE 112/158 single nucleotide polymorphisms (which determine the E2, E3, and E4 isoforms) was performed through PCR followed by pyrosequencing. PCR was used for amplification with the following primer pairs: ApoE 112: forward, 5′-ACGGCTGTCCAAGGAGCTG-3′, and reverse, biotin 5′-CACCTCGCCGCGGTACTG-3′; and ApoE 158: forward, 5′-CTCCGCGATGCCGATGAC-3′, and reverse, biotin 5′-CCCCGGCCTGGTACACTG-3′. Genomic DNA (250–500 ng) was amplified with 20 pm of each primer, 1×PCR buffer kit (Qiagen), 0.4 mm dNTP, 1.0 mm MgCl2, DMSO, and 0.01 U of Qiagen Taq polymerase. A Biometra TProfessional Basic Thermocycler was used for amplification with the following conditions for 35 cycles: 30 s at 95°C, 30 s at 58.6°C, or 58.1°C for ApoE 112 or 158 respectively, and 1 min at 72°C. These 35 amplification cycles were preceded by a 2 min hot start at 95°C and were followed by a final 4 min extension to the last cycle at 72°C. PCR products were visualized on a 1.2% agarose gel. The polymorphisms were subsequently determined via an established pyrosequencing protocol with oligo sequencing for ApoE112 (5′-CGGACATGGAGGACG-3′) and ApoE 158 (5′-CGATGACCTGCAGAAG-3′). The analyzed sequence was as follows: TGT/CGCGGCCGCCT for ApoE112 and T/CGCCTGGCAG for ApoE158.
Quebec Founder Population cohort: Autopsy-confirmed case/control subjects
The QFP cohort is a Canadian population that descends from French settlers who founded Nouvelle France in the 17th and 18th centuries. A founder effect was created in this population due to the migration and isolated nature of the settlements, resulting in less genetic heterogeneity, large linkage disequilibrium blocks and low genetic noise. QFP human brain tissue was obtained from the Douglas Bell Canada Brain Bank. This study had the approval of the Douglas Research Center institutional review board and was performed in conformity with the Code of Ethics of the World Medical Association (McGill/Douglas ethic approvals A05-B16-11B and IUSMD-02-34). The histopathological diagnosis of AD followed the NINCDS-ADRDA (National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association) criteria [22].
DNA extraction and genotyping
APOE4 allele determination was performed on brain tissue samples with DNeasy tissue extraction kit (Qiagen Hilden, Germany) and the pyrosequencing protocol described above.
RNA extraction and quality control
RNA was extracted from frontal cortex tissues (n = 88) using the Maxwell® 16 Tissue LEV Total RNA Purification Kit (Promega, WI, USA) on a Maxwell® 16 LEV Instrument (Promega, WI, USA). Then, cDNA was obtained by reverse transcription on a Multigene thermocycler (Labnet International Inc.) using the high-capacity cDNA RT kit (Applied Biosystems, CA, USA) and 200 ng of total RNA. The purity and integrity of RNA samples were estimated using the ratio of absorbance values at 260 and 280 nm evaluated on a Biotek Synergy H1 reader (Fisher Scientific, ON, Canada), and the RNA integrity number (RIN) determined with a Bio-Rad’s Experion instrument (Bio-Rad, CA, USA). The ratios of absorbance were all over 1.5, while RINs ranged from 2 to 8.4, with 84% of samples over 5, the cutoff value representing very good total RNA quality [23] for microarray methodology.
Microarray
The purity and integrity of extracted RNA were estimated using, respectively, the ratio of absorbance values at 260 nm and 280 nm evaluated on a Biotek Synergy H1 reader (Fisher Scientific, ON, Canada), and the RNA integrity number (RIN) determined with a Bio-Rad’s Experion instrument (Bio-Rad, CA, USA) before processing with the Applied Biosystem Clariom D microarray according to the manufacturer’s protocols. Gene-level expressions of APOE, CLU, APOD, APOB and CNTN5 transcripts in the frontal cortex were estimated using the transcriptome analysis console (Applied Biosystem, USA) following standardization protocols.
Animal studies
Animals
All animal procedures were performed in conformity with the Canadian Guidelines for Use and Care of Laboratory Animals and were approved by the McGill University Animal Care Committee (approval DOUG-10032). Animals used were 2–3-month-old male C57BL/6J wild-type mice. All mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA), housed individually, and fed standard laboratory chow ad libitum. Animals were kept in a 12-h-light-dark cycle, with light onset at 07 : 00 and offset at 19 : 00.
Unilateral entorhinal cortex lesions
Unilateral electrolytic lesions were performed on the entorhinal cortex of adult mice as previously described [24]. Animals were anesthetized with isoflurane and placed in a stereotaxic apparatus in a flat skull position. Lesion coordinates were determined from Lambda in the following positions: 1) [AP: 0 mm], [L: –3 mm], [DV: –3 mm, –4 mm]; 2) [AP: 0 mm], [L: –3.5 mm], [DV: –3 mm, –4 mm]; 3) [AP: +0.5 mm], [L: –4 mm], [DV: –3 mm, –4 mm]; 4) [AP: +1 mm], [L: –4 mm], [DV: –3 mm, –4 mm]. A current of 1 mA was applied at each coordinate for 10 seconds. For sham-operated mice, which are used as the control group, the same steps were followed, but the electrode was lowered only 1 mm, and no current was applied. After surgery, subcutaneous physiological saline was given to prevent dehydration and animals were nursed throughout their recovery. Lesioned mice were sacrificed at 2, 7, 14, 21, and 40 days post-lesion (DPL) and sham-lesioned mice were sacrificed on the same day as surgery. Six mice were sacrificed at each time point. Animals were decapitated, the brain was quickly removed, hippocampi contralateral and ipsilateral to EC lesion was dissected and stored at –80°C.
RNA extraction and quality assessment
RNeasy lipid tissue mini kit (Qiagen, Hilden, Germany) was used for RNA extraction. RNA quality was assessed at McGill Genome Centre. All RNA samples had RIN > 7.8 and 260/280 ratios > 2.1.
Hippocampal CNTN5, APOE, CLU (APOJ), APOB, and APOD mRNA levels
Hippocampal mRNA levels of CNTN5, APOE, APOJ, APOB and APOD were measured at different time points (n = 6 animals per time point) by the McGill Genome Centre, using the Mouse Clariomtrademark D genechip assay (Affymetrix, Santa Clara, CA, USA).
Statistical analysis
The differences in population distribution were assessed using the chi-square test. The difference in age between AD subjects and controls was measured using the Student’s t-test. The correlational analyses between contactin 5 and apolipoproteins or cholesterol were performed in the plasma and CSF using linear regression models corrected for age, sex and APOE4. The correlation between contactin 5 mRNA and apolipoprotein mRNA in the frontal cortex was measured using linear regression models. The Kruskal-Wallis test was used to evaluate the difference in mean mRNA levels between different days post-lesion in the lesioned mouse model. Significance level was considered at p < 0.05.
RESULTS
Demographics
Table 1 summarizes the demographic characteristics of the cohorts used. In the QFP cohort there were no differences in the proportion of males and females or in age between diagnostic groups, but AD subjects were more likely to be APOE ɛ4 positive compared to controls (p = 0.01).
PREVENT-AD and QFP demographics
APOE, apolipoprotein E; PREVENT-AD, Pre-symptomatic Evaluation of Experimental and Novel Treatment for Alzheimer’s disease; QFP, Quebec Founder Population.
Associations in the CSF
Contactin 5 was found to positively correlate with apolipoproteins B (r2 = 0.62, p = 5.4×10–8), D (r2 = 0.302 p = 1.86×10–4), E (r2 = 0.370, p = 2.92×10–9), and J (r2 = 0.386, p = 2.65×10–9) in the CSF of cognitively unimpaired subjects from the PREVENT-AD cohort (Fig. 1). In contrast, CSF apolipoproteins A1, A2, C1, C3, and H (all with peripheral origin) displayed no association with contactin 5 (Fig. 1).
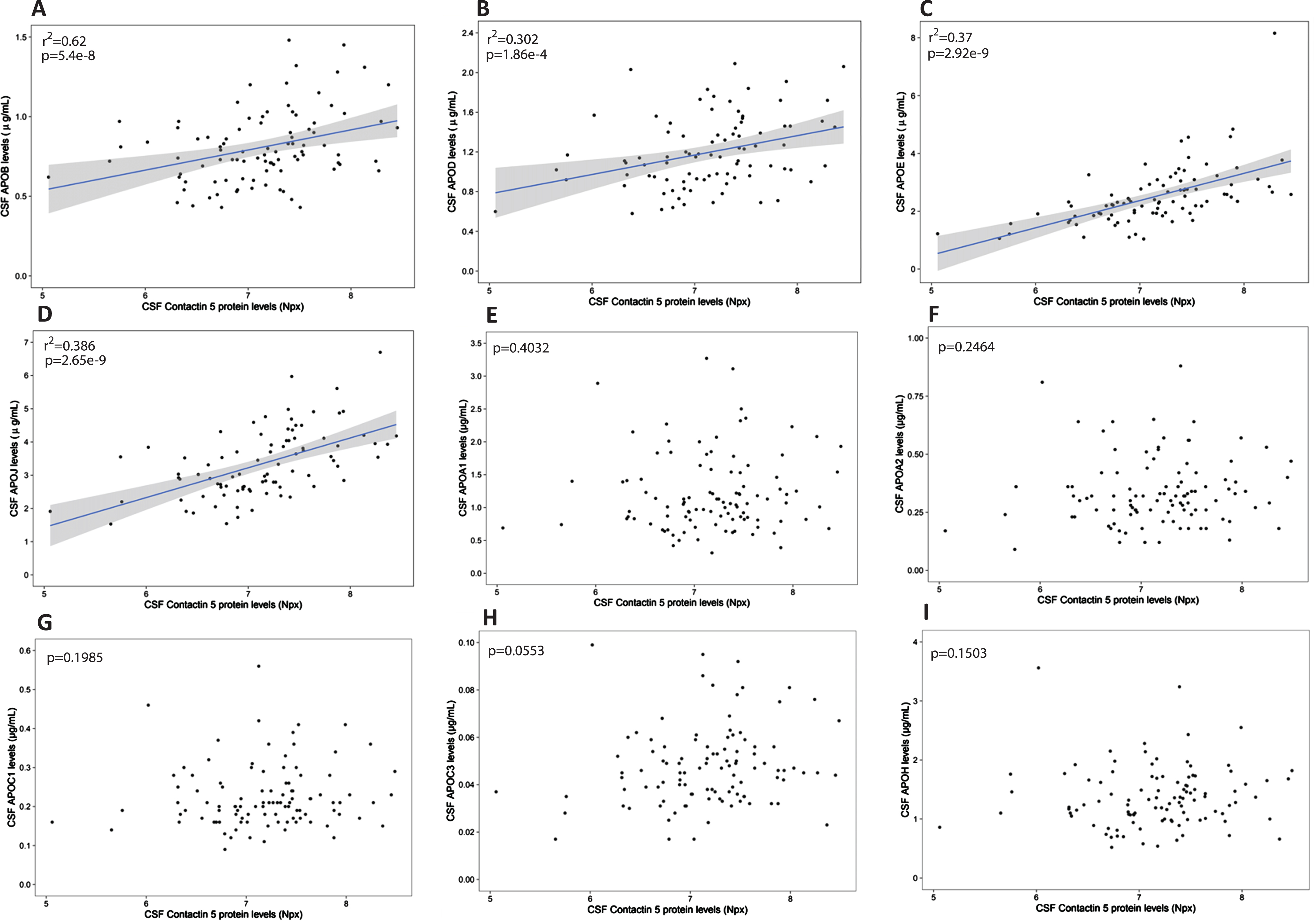
Association between CSF Contactin 5 and apolipoproteins. Contactin 5 protein was measured in the CSF using Olink’s proximity extension assay. Apolipoproteins were measured in the CSF using Luminex. Significant linear regressions are represented with a gray confidence region of the fitted line. Individual R squares and p values are shown in the top left corners of each figure.
CSF cholesterol concentration (but not 25-hydroxycholesterol) positively correlated with contactin 5 concentration (r2 = 0.1352, p = 0.0096, Fig. 2).
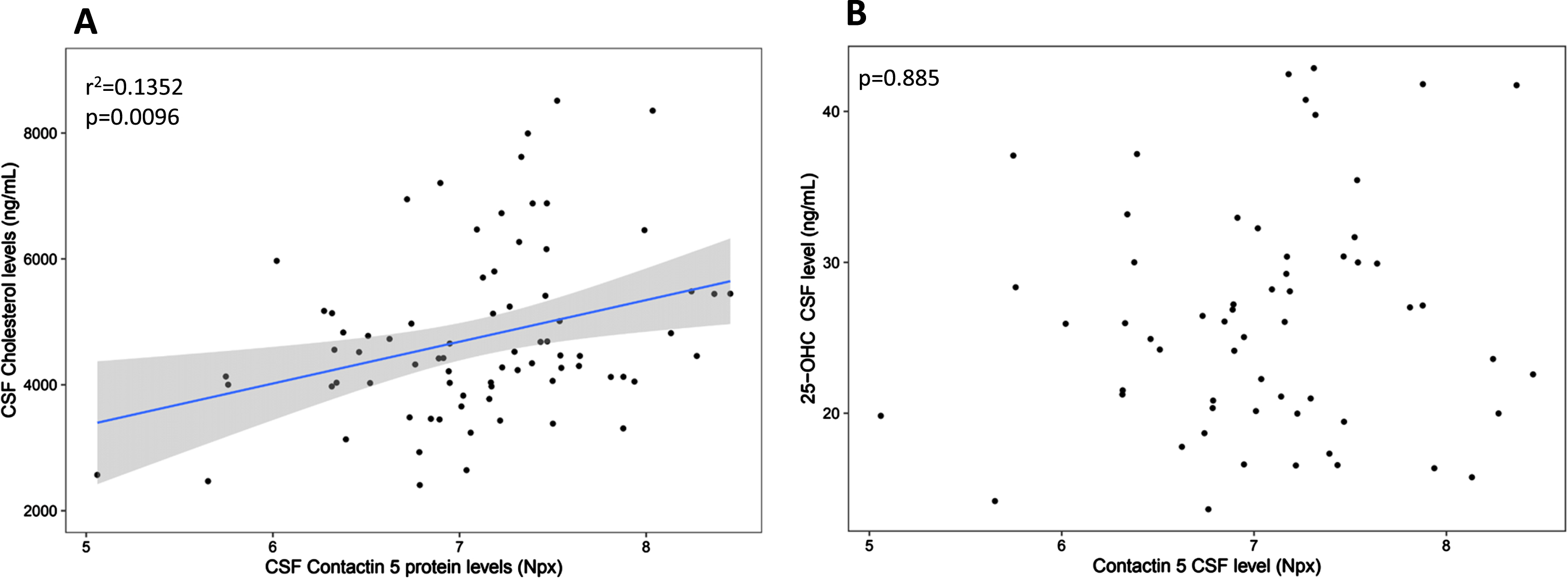
Association between CSF Contactin 5 and Cholesterol and between CSF Contactin 5 and 25-OHC. Contactin 5 protein was measured in the CSF using Olink’s proximity extension assay. Cholesterol and 25-OHC were measured using liquid chromatography mass spectrometry. Significant linear regressions are represented with a gray confidence region of the fitted line. Individual R squares and p values are shown in the top left corners of each figure.
Stratification by APOE genotype (E4 positive versus E4 negative) was used to assess the effect of the most important genetic risk factor for sporadic AD in the PREVENT-AD cohort (Fig. 3). In APOE4 positive subjects, CSF contactin 5 positively correlated with apolipoproteins B (r2 = 0.431 p = 0.00005), D (r2 = 0.323 p = 0.04), E (r2 = 0.620 p = 0.0000003), and J (r2 = 0.466 p = 0.000374). In APOE4 negative subjects, contactin 5 positively associated with apolipoproteins B (r2 = 0.168 p = 0.003), D (r2 = 0.311 p = 0.002), E (r2 = 0.326 p = 0.00002), and J (r2 = 0.437 p = 0.0000004) (Fig. 3).
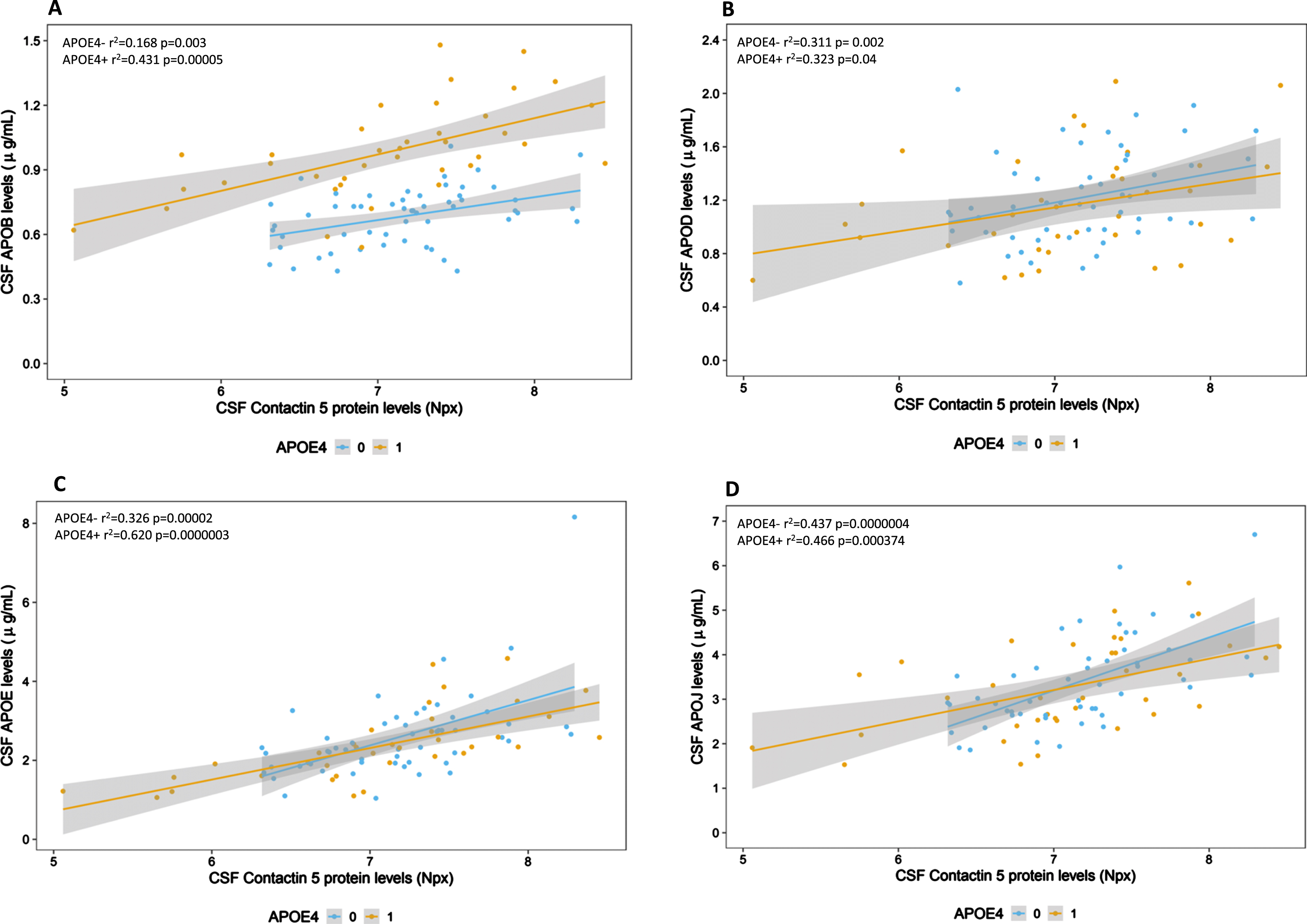
Association between CSF Contactin 5 and apolipoproteins divided by the presence and absence of the APOE4 allele. Contactin 5 protein was measured in the CSF using Olink’s proximity extension assay. Apolipoproteins were measured using Luminex. Significant linear regressions are represented with a gray confidence region of the fitted line. Individual R squares and p values are shown in the top left corners of each figure.
A noteworthy observation is that the trajectory of CSF apoB levels show parallel rather than overlapping trajectories in those who are E4-positive compared to those who are E4-negative (Fig. 3A). Other apolipoproteins (D, E, and J) display parallel and overlapping slopes (Fig. 3B–D): suggesting different lipoprotein compartments from that of apoB.
Associations in the plasma
In the plasma, contactin 5 positively correlated with peripheral cholesterol (r2 = 0.1934, p = 0.02), HDL (r2 = 0.2339, p = 0.0143), and LDL (r2 = 0.1267, p = 0.0121) concentrations (Fig. 4). However, in contrast to the CSF, there were no significant correlations between contactin 5 and apolipoproteins B, D, E, or J in the plasma compartment (not shown).
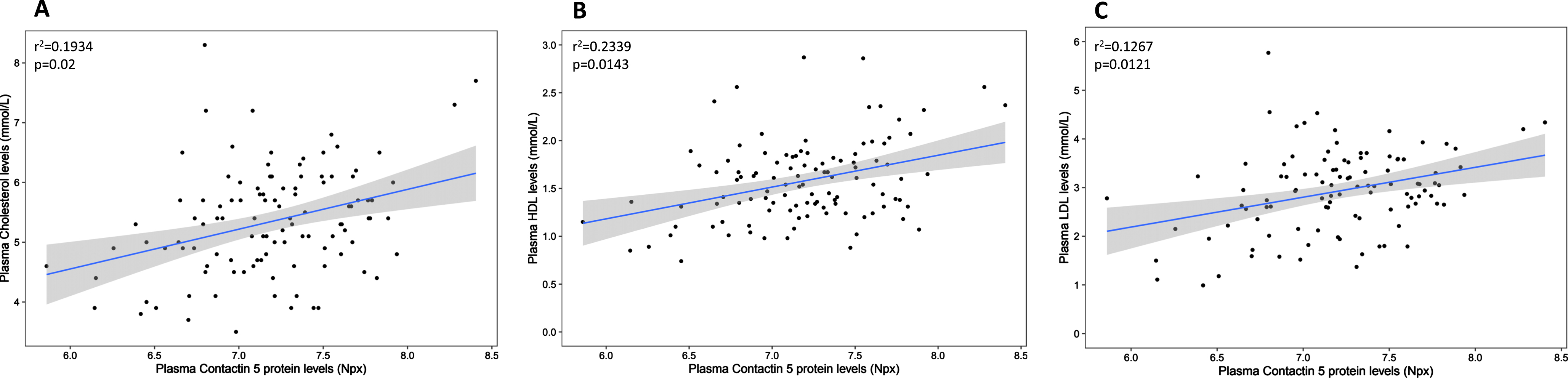
Association between plasma Contactin 5 and cholesterol. Contactin 5 protein was measured in the plasma using Olink’s proximity extension assay. Cholesterol was measured using liquid chromatography mass spectrometry (LCMS/MS). Significant linear regressions are represented with a gray confidence region of the fitted line. Individual R squares and p values are shown in the top left corners of each figure.
Gene expression in the frontal cortex in autopsy-confirmed AD and control cases
Figure 5 contrasts levels of APOE, APOB, APOD, and CLU mRNAs as a function of CNTN5 mRNA prevalence in the frontal cortex of AD and control cases from the QFP cohort. There are no significant differences in cortical CNTN5 gene expression between AD and controls when adjusted for APOE4, sex and age (Fig. 5A). Significant negative correlations were seen between CNTN5, APOB (r2 = 0.15, p = 0.034) and APOE (r2 = 0.192, p = 0.015) mRNA levels in the control group but not in the AD cases (Fig. 5B).
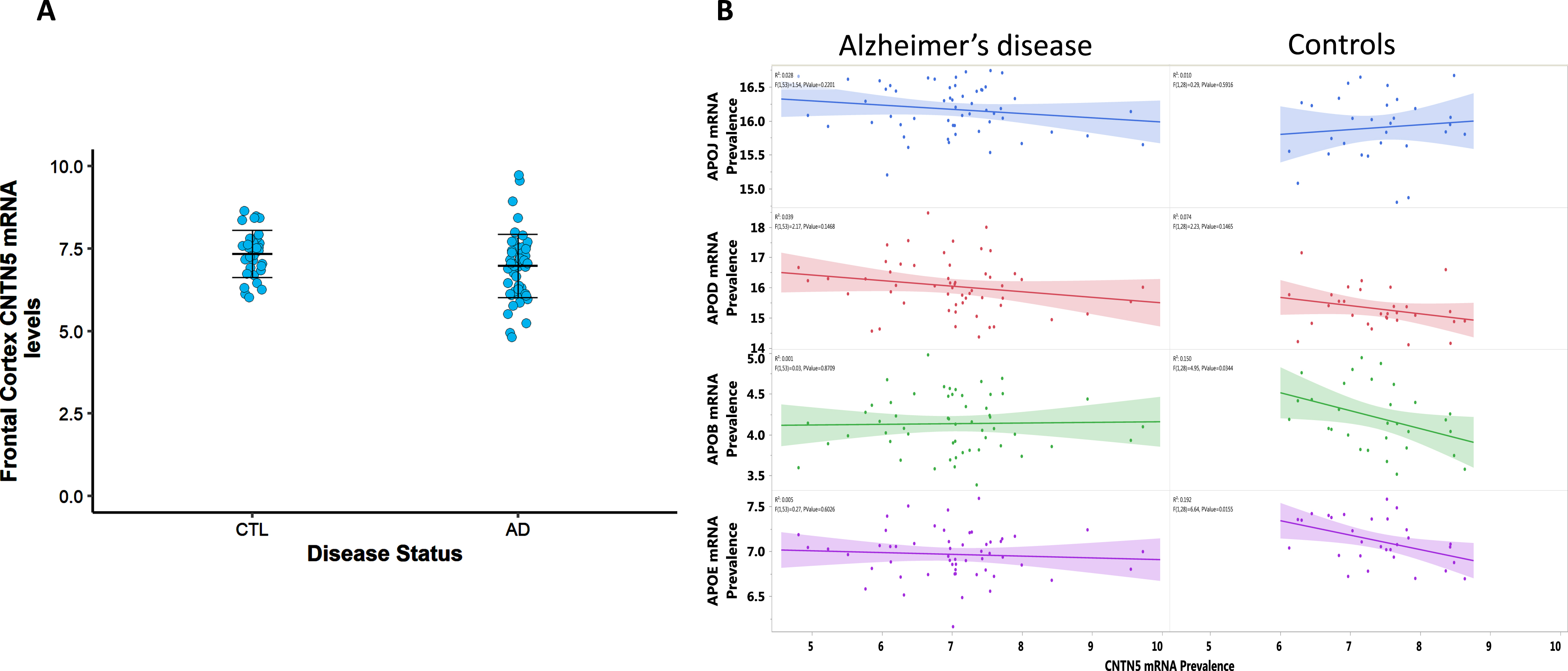
A) Contactin 5 mRNA levels in the frontal cortex of AD (n = 57) and controls (n = 31). mRNA was measured using qRT-PCR. p > 0.05. The demarcated lines represent the mean and SD. B) Association between contactin 5 and apolipoproteins mRNA levels in the frontal cortex of AD subjects and controls. mRNA was measured using qRT-PCR. Significant linear regressions are represented with a colored confidence region of the fitted line. Individual R squares and p values are shown in the top left corners of each figure.
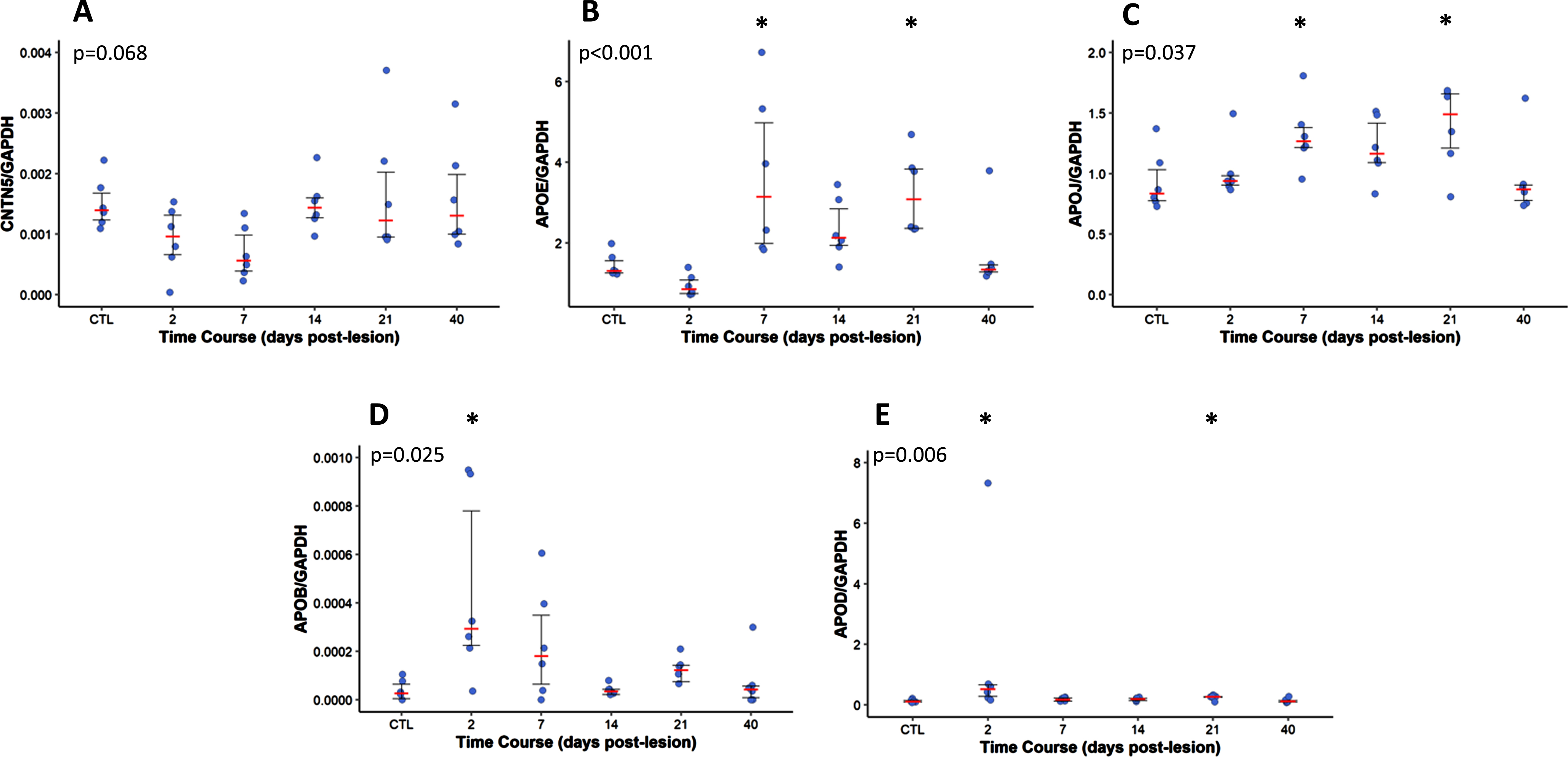
Gene expression of Contactin 5 and apolipoproteins in a mouse model of entorhinal cortex lesion (ECL). The dots represent the values of either an apolipoprotein or contactin 5 divided by GAPDH (n = 6 animals per time point). The demarcated lines represent the median and the interquartile range. The p-value for each protein is on the top left corner of each figure. *p < 0.05 as compared to day 0.
Gene expression in a mouse model of hippocampal deafferentation and reinnervation
Figure 6 illustrates the time course analysis of the hippocampal mRNA prevalence for cntn5, apoe, clu (apoJ), apob, and apod as a function of deafferentation (0–10 days) and the ensuing reinnervation (10–40 days) process. The ipsilateral modifications are being contrasted with the outcomes from the control group (sham mice). A transient decrease in cntn5 mRNA levels (trends only, p = 0.068) can be seen 7 days after the lesioning, which coincides with the peak of neuronal deafferentation in the hippocampus. In contrast, apoe and apoj gene expression increased at 7 DPL (p < 0.05) in the early phase of the reinnervation process and remained elevated during the reinnervation phase until 21 DPL (p < 0.05). There were transient increases of apob and apod at 2DPL (p = 0.025 and p = 0.006, respectively), which is indicative of the initial stage of the deafferentation process (p < 0.05).
DISCUSSION
We studied the interaction between contactin 5, apolipoproteins, and cholesterol in elderly individuals who were cognitively unimpaired but carrying a greater risk of developing AD. We observed extensive interactions between these molecules in both CSF and plasma. The recent identification of several genetic risk factors for sporadic AD by GWAS that belong to key regulatory genes involved in cholesterol metabolism such as APOE4 and CLU (lipoprotein-mediated cholesterol transport), PICALM, SORL1, and BIN1 (lipoproteins internalization), ABCA1 and ABCA7 (intracellular cholesterol transport and mobilization) [25–27] further emphasize the importance of such interactions in the mature and aging brains.
Indeed, all CNS-relevant apolipoproteins shown here to interact with contactin 5 had been previously associated with different aspects of AD pathology. For example, apoE is actively involved in cholesterol transport and Aβ catabolism, whereas the APOE ɛ4 variant shows lower affinity for lipids and leads to less effective lipoprotein-mediated cholesterol and amyloid transport and increased Aβ production combined to decreased Aβ clearance [28, 29]. In addition, APOE and APOA1 were shown to independently protect from amyloid precursor protein carboxy terminal fragment-associated cytotoxicity [30]. CLU (apoJ), another important genetic risk factor for AD is a major component of lipoprotein complexes (HDL-like) and has also been involved in amyloid transport and catabolism [31, 32]. Similarly, apoB and apoD participate in cholesterol transport as minor components of HDL-like particles, and both have been found to be elevated in the CSF of individuals with AD and to markedly associate with tau pathology [5, 7].
Variants in the CNTN5 gene have been associated with increased risk for neurodevelopmental disorders such as attention deficit hyperactivity disorder [33] and autism [34, 35]. CNTN5 genetic variants have been associated with increased risk for AD (rs1461684 and rs10501927) [19, 26] and a faster rate of progression (rs1461684) [19] in the pre-symptomatic phase of the disease. The level of contactin 5 protein is elevated in the CSF of those who are “at-risk” but have not yet experienced cognitive impairment, while decreased levels have been reported in MCI and AD subjects [19], similar to other contactin species [36]. Contactin 5 is also associated with CSF tau, phospho-tau levels, and synaptic markers, and to a lesser extent, to amyloid [19].
Contactin 5 is a neuronal membrane protein that acts during neurodevelopment on neuronal migration, axonal guidance, myelin formation and synaptogenesis [37, 38]. It is particularly involved in axonal arborization, synaptic formation and remodeling [17, 18]. In the mice, cntn5 expression pattern included strong expression in the cerebral cortex in layers II–V, hippocampus and mammillary bodies in addition to previously described brain nuclei of the auditory pathway and the dorsal thalamus [39]. Deletion of the cntn5 gene in mice leads to defects in the cortical and subcortical auditory pathways and loss of presynaptic inhibitory boutons in multiple brain areas [40, 41]. The association between contactin 5 and apolipoproteins seen in the CSF of these “at-risk” subjects for AD is very significant because not only both families of proteins have been involved in the pathology of AD, but they share complementary functions as they are both involved in synaptic maintenance and remodeling in the CNS.
Synaptic dysfunction and axonal loss are important early events in AD preceding cognitive decline [42]. Given the well-established mechanisms of action of contactin 5 and apolipoproteins in synaptic physiology, it is conceivable that in response to early synaptic damage, lipophilic contactin 5 is released in the extracellular space where it is taken up by lipid-rich lipoproteins and transported to target cells in the CNS and/or eliminated from the CNS. This is consistent with the finding that contactin 5 is also positively associated with cholesterol, HDL and LDL in the plasma as well as with cholesterol in the CSF. The soluble form of contactin-2, a contactin 5 analog, acts as a guiding molecule for the outgrowth of neurites and plays a role in axon extension initiation, axonal guidance and fasciculation [43, 44]. It is thus conceivable that contactin 5, which also acts as a scaffold on inter-neurons where dendrites of direction-selective neuronal cells can fasciculate, requires both secretion and the presence of lipid-rich HDLs complex to provide reinnervating neurons with the necessary lipids for effective neurite outgrowth and axonal extension [41, 46].
To better understand the possible origin of the association between contactin 5 and apolipoproteins in the CNS, we measured and contrasted their gene expression in the frontal cortex of autopsy-confirmed AD and control subjects. mRNA levels of CNTN5 negatively associate with APOB and APOE transcripts in control subjects, but not in AD. These findings show that the associations between these proteins in the CSF compartment are not due to changes in gene expression but are more likely a consequence of the neuronal damage that occurs in the early phase of the neurodegenerative process.
To further investigate the role of contactin 5 and apolipoproteins during axonal sprouting and terminal proliferation in the mature CNS, we measured the gene expression of contactin 5 and apolipoproteins in a well-established model of rodent hippocampal deafferentation/reinnervation: the entorhinal cortex lesioning paradigm [47]. In this model, using wild-type animals, it is possible to examine without the interference of amyloid and tau pathology, the alterations in gene expression that occur in the hippocampus in response to a lesion to the entorhinal cortex and the resulting loss of input due to degeneration of the perforant pathway. It gave us the opportunity to observe how the gene expression alterations of these potential markers reacted to the synaptic loss that occurred in the initial 10 days and the consecutive terminal and synaptic restructuring that occurred in the 14–42 days window after the lesion.
Results show that apob and apod mRNA levels are increased early on during the deafferentation phase (2 DPL) while the gene expression of the major CNS cholesterol transporters apoe and apoj increases in the late phase of the deafferentation process and peak during the early stage of the reinnervation process, up to 21 DPL. This is consistent with previous literature that shows peak elevation of apoE mRNA levels between 7 and 14 DPL, during the early phase of the reinnervation process, when terminal and synaptic remodeling begins [47, 48]. Contactin 5 mRNA levels display a time-dependent reduction in the 0–7 DPL window (p = 0.06 at 7 DPL), with levels returning to normal after day 14. This transient reduction is identical to the time-course alterations reported for other synaptic markers such as GAP-43, synaptophysin and SNAP-25 [47, 50]. These results suggest that the gene expression of apolipoproteins (and possibly contactin 5) does change in response to synaptic deafferentation, but the elevation at different time-points suggests each protein plays a different role in the deafferentation/reinnervation process.
In summary, we show that extensive interactions exist between apolipoproteins and contactin 5 in asymptomatic subjects at high-risk of AD. Additionally, we show that following a lesion to the entorhinal cortex in a mouse model, significant alterations in gene expression of apolipoproteins occur at key moments in the reinnervating hippocampus. The precise role of contactin 5 and apolipoproteins in the pathophysiology of AD is not completely understood at this time, but these results suggest a possible active role in axonal, terminal and synaptic remodeling in response to entorhinal cortex damage due to experimental lesions or, to AD pathology in humans.
Follow-up studies looking into the biochemistry of these proteins throughout the spectrum of AD pathology would be important to better understand the pathophysiology of the disease and more specifically how the brain reacts to this type of pathological damage.
Furthermore, in this initial work into the role of contactin 5 and apolipoproteins following neuronal damage, we aimed to investigate the earliest changes in gene expression. In the animal model of hippocampal deafferentation, that meant focusing on mRNA levels. However, to fully elucidate the role of these molecules on neuronal repair, it is crucial to also understand the changes that occur in protein level and function. Work is currently underway to investigate protein changes in the hippocampus in response to entorhinal cortex deafferentation.
AUTHOR CONTRIBUTIONS
Marina Tedeschi Dauar (Conceptualization; Data curation; Formal analysis; Methodology; Writing – original draft; Writing – review & editing); Cynthia Picard (Data curation; Formal analysis; Investigation; Methodology; Writing – review & editing); Anne Labonté (Data curation; Formal analysis; Investigation; Methodology; Writing – review & editing); John Breitner (Funding acquisition; Investigation; Methodology; Project administration; Resources; Writing – review & editing); Pedro Rosa Neto (Data curation; Investigation; Writing – review & editing); Sylvia Villeneuve (Funding acquisition; Investigation; Methodology; Resources; Writing – review & editing); Judes Poirier (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Writing – original draft; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
The authors wish to thank Jennifer Tremblay-Mercier, Doris Dea, and Louise Théroux for their individual contribution at different stages of the project. The authors would also like to thank Dr. Naguib Mechawar at the Douglas Institute/Bell Canada Brain Bank for providing human brain tissues from the Québec Founding Population. The authors also wish to acknowledge the PREVENT-AD participants and their families as well as all the PREVENT-AD team members for their time and dedication.
FUNDING
Marina Tedeschi Dauar is supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) – Brasil – (88881.128434/2016-01). Dr. Poirier is supported by the Fonds de la Recherche en Santé du Québec (FRSQ), the Canadian Institute for Health Research (CIHR# PJT 153287), and the J.L. Levesque Foundation.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
Data used in preparation of this article were obtained from the program of PRe-symptomatic EValuation of Novel or Experimental Treatments for Alzheimer’s Disease (PREVENT-AD) at the Centre for Studies on Prevention of Alzheimer’s Disease (StoP-AD), Douglas Mental Health University Institute Research Center https://preventad.loris.ca/main.php). A complete listing of the PREVENT-AD Research Group can be found at: ![]() .
.
The PREVENT-AD cohort data is open access and available online through data releases following requests https://prevent-alzheimer.net/ and ![]() ).
).
Animal and human pathological data supporting the findings of this study are available on request from the corresponding author.