
Abstract
Background:
Previous reports have demonstrated post-operative dementia and Alzheimer’s disease (AD), and increased amyloid-β levels and tau hyperphosphorylation have been observed in animal models post-anesthesia.
Objective:
After surgical interventions, loss in memory has been observed that has been found linked with genes modulated after anesthesia. Present study aimed to study molecular pattern present in genes modulated post anesthesia and involved in characters progressing towards AD.
Methods:
In the present study, 17 transcript variants belonging to eight genes, which have been found to modulate post-anesthesia and contribute to AD progression, were envisaged for their compositional features, molecular patterns, and codon and codon context-associated studies.
Results:
The sequences’ composition was G/C rich, influencing dinucleotide preference, codon preference, codon usage, and codon context. The G/C nucleotides being highly occurring nucleotides, CpGdinucleotides were also preferred; however, CpG was highly disfavored at p3-1 at the codon junction. The nucleotide composition of Cytosine exhibited a unique feature, and unlike other nucleotides, it did not correlate with codon bias. Contrarily, it correlated with the sequence lengths. The sequences were leucine-rich, and multiple leucine repeats were present, exhibiting the functional role of neuroprotection from neuroinflammation post-anesthesia.
Conclusions:
The analysis pave the way to elucidate unique molecular patterns in genes modulated during anesthetic treatment and might help ameliorate the ill effects of anesthetics in the future.
Keywords
INTRODUCTION
In 1955, the initial description of cognitive deterioration and anesthesia and surgery in older people was described. In the study of Rundshagen and colleagues in 2014, 12% of the patients above 60 years of age suffered postoperative cognitive dysfunction three months post-surgery [1]. The pathological hallmark of Alzheimer’s disease (AD) is amyloid-β (Aβ) plaque and tau protein hyperphosphorylation. Multiple factors are risk factors for AD, including elderly age, family history, trauma, and genetic mutations [2]. Among genetic causes, mutations in presenelin 1 and presenelin 2 genes account for 70% and 20–25% of the cases of early-onset AD, respectively [3]. Another gene is APOE, for which variant 4 is responsible for the early onset of AD [4]. Commonly used inhalation anesthetics are isoflurane, sevoflurane, and desflurane, and they have been demonstrated to induce cellular apoptosis and increase Aβ generation [5]. Anesthetic 2.1% sevoflurane for six hours, can induce caspase-3 activation and increased Aβ levels with neurotoxic effects on the brain [6]. Anesthesia induces hypothermia, accelerating tau pathology in vivo irrespective of the anesthetic used [7]. Also, a link between the dose and duration of anesthetic and neurodegenerative changes has been observed [8].
Mice models have demonstrated that anesthesia induces neuroinflammatory genes and is implicated in dementia progression. LRP1 removes Aβ peptides from brain interstitial fluid to blood to eliminate them through the hepatic system [9]. APOE and LRP1 have been found as links between anesthesia and AD [10], where ApoE4 is increased while LRP1 levels are decreased. Genes CD68, Aldh1l1, Cx3cl1, APOE4, Lrp1, TREM2, CLU, and ABCA7, are found differentially expressed in the anesthetized animal model [10]. Considering the importance of these genes in triggering AD and latent tau protein aggregation post-anesthesia, we were tempted to study the codon usage pattern and molecular signature investigation for these proteins. The function of these genes concerning AD development is given in Table 1. Codon usage and codon context of any sequence determine the expression level of the gene and are correlated to various parameters like protein properties, protein folding, expression rates, and level of protein expression. Thus, we envisaged these genes for codon usage, codon context determination, and other molecular patterns to envisage molecular signatures in the sequences.
Genes triggered post-anesthetic treatment and having a role in triggering Alzheimer’s disease (AD) and latent tau protein aggregation
MATERIALS AND METHODS
Sequence retrieval and nucleotide compositional analysis
The transcript sequences for eight genes mentioned in Table 1 were retrieved from the National Center for Biotechnology Information in FASTA format. A total of 17 coding transcripts were obtained for 08 genes. The sequences included in the study were in the reading frame and started with the start codon and ended with the termination codon and did not contain any ambiguous codon.
Nucleotide composition is a feature that influences codon usage [22], protein properties [23], gene expression [24], and evolutionary forces like mutation [25]. In the present study, we envisaged overall nucleotide composition and composition at the third nucleotide position (percent composition of A, T, C, and G nucleotide and percent composition of A3, T3, C3, and G3 nucleotides, respectively). Percent GC composition at all three codon positions was also determined. The obtained results were used for parity analysis, neutrality plot, and correlation analysis.
Dinucleotide odds ratio
Out of four nucleotides, combinations of sixteen dinucleotides are possible. The ratio of observed to the expected value of dinucleotide frequency is called the odds ratio. The odds ratio below 0.78 and above 1.23 are called underrepresentation and overrepresentation, respectively [26]. The anaconda2 tool by Moura et al. (2005) was used [27] to derive the odds ratio at codon junction. The overall dinucleotide odds ratio was calculated using CodonW 1.4.2 package.
The relative synonymous codon usage (RSCU) analysis
RSCU is a measure to know codon bias and suggests the relative frequency of codons [28]. In the present study, we studied the RSCU value of 59 codons (Methionine and tryptophan were excluded since these are encoded by a single codon, and stop codons were also excluded). RSCU value is zero when some codon is absent in sequence or is 1 when there is no bias in the codon usage. The maximum possible value for RSCU can be 6 in the case of the six-codon family, where a single amino acid is encoded by six codons, and out of six codons, only one is used [29]. RSCU values were calculated using a tool developed by Puigbò et al. (2008) [30] and available at http://genomes.urv.es/CAIcal/.
Synonymous codon usage order (SCUO)
SCUO is a directional measure, a parameter to determine the codon bias. For measuring SCUO, first, the entropy of the i-th amino acid was calculated as Hi, where it is between 1 and 1 for the i-th codon, between 1–6 for leucine, between 1–2 for tyrosine and likewise for other degenerate amino acids. If the i-th amino acid is used randomly, then the maximum entropy of the i-th amino acid will be represented as Himax. Followed by calculated maximum entropy SCUO can be calculated for individual i-th amino acid. Average SCUO is the measure of codon bias. The SCUO value was calculated using the formula of Angellotti et al. (2007) [31]. Its values range between 0 and 1, where 0 presents no bias to 1, which is the highest bias. It is an entropy-based measure.
Neutrality plot
We plotted a regression plot between GC content at the third codon position (% GC3) and the average content of GC present at the first and second codon position (% GC12). % GC3 exhibits the selection forces since most nucleotide changes at the third codon position are neutral. GC content at positions first and second is subjected to functional constraints against change, and mutations here lead to changes in amino acid [32]. Here, % GC3 is plotted at the X axis, and Y is plotted at the Y axis. The regression coefficient near 1 shows that mutation force is solely responsible for codon usage and vice versa. A value near zero indicates dominance of the selection force.
Parity rule analysis
A parity plot is constructed between AT bias ((A3/(A3 + T3)) and GC bias ((G3/(G3 + C3)) at the third codon position. At the center of the plot, where the value is 0.5, the parity plot indicates no bias, and mutation and selection play equal roles in shaping codon bias. Otherwise, selection remains the main evolutionary force influencing codon usage [33].
Protein properties
Proteins possess various physiochemical properties like isoelectric point, instability index, aliphatic index, GRAVY and AROMA, hydrophobicity, and the frequency of acidic, basic, and neutral amino acids [34]. The isoelectric point of a protein is a pH at which the net charge on the protein is zero, and after applying the electric field, it does not move towards any charged field. The pH required for optimal stability and biological activity differs according to the isoelectric point [35]. The instability index is a measure that speculates on the stability of a protein in vitro. An instability index less than zero indicates a possible stability [36]. Aliphatic side chains of a protein occupy some space in the dimension, and the relative volume of this space is called the Aliphatic index (AI) and confers roles in the thermal stability of protein [37]. GRAVY is the Grand Average of Hydropathy and is suggestive of the hydrophobicity of a protein and is obtained by dividing the sum of hydropathy values of individual amino acids of a protein by the length of a protein. Positive GRAVY values suggest the protein is hydrophobic, while negative values indicate a hydrophilic nature [38] and it ranges between –2 and+2 [39]. AROMA (aromaticity) is the frequency of Phe, Tyr, and Trp aromatic amino acids, and the interactions between aromatic amino acids influence many biological processes as an integral part of many interactions within protein and DNA and protein [40]. Hydrophobicity controls the three-dimensional structure, and proper folding of the protein and globular structure of most of the proteins is due to hydrophobicity. Changes in the surface hydrophobicity might change the molecular characteristics of a protein [41]. Replacement of basic amino acids with the acidic amino acids in the peptide chain enhances the solubility of the protein [42]. While basic amino acids help in salt formation and therefore, the solubility of Indomethacin (a nonsteroidal anti-inflammatory drug) has been improved [43]. Codon usage correlates to the protein properties [44]. Hence, we investigated nine protein properties in the present study for their possible linkage with the codon bias. Protein properties were calculated using webtool available at link https://web.expasy.org/protparam/.
Codon context analysis
Similar to codon bias, where certain codons are preferred over others, bias is found in codon pairs, where the presence of certain codon pairs are favored, called codon pair bias or codon context [45, 46]. Condon context was determined using the Anaconda2 tool, used for statistical analysis of the contingency table and tells if the context is significantly biased and provides information regarding preferred and rejected pairs of codons [46].
Principal Component Analysis (PCA)
PCA is an analysis to determine major trends if a large number of variables are present. Using PAST4 software, PCA analysis was done. There were 59 codons corresponding to 59 variables. Here, three stop codons and Trp and Met were excluded. The RSCU values are transformed into uncorrelated variables. A PCA plot was generated using the first two axes.
RESULTS
The sequences are GC rich
Nucleotide composition affects various properties of sequences like codon bias, gene expression [47], length of transcript [48], and protein properties [49]. The average percent composition of T was the lowest (10.39±3.33), while % G was the highest (31.96±5.78). At third codon position, % A3 was least (17.02±2.73) and % G3 was highest (39.29±11.22). The envisaged sequences were C rich (34.29±3.11%) while % G composition is slightly less (31.62±5.78%). However, the same is not true for the third codon position where both the % C and % G were almost similar, 36.74±4% and 36.1±11.22%.
CpG dinucleotide is overrepresnted in few sequences
Codon TpT and TpA were underrepresented (odds ratio below 0.78) in all the envisaged sequences. In all the five APOE transcripts, GpC and GpG dinucleotides were highly represented with odds ratio≥2.3 and≥2.0 for GpC and GpG, respectively. CpC presented overrepresentation (RSCU≥2.0) in CD68 and CXCL1 transcripts (Fig. 1). Codon TpA was underrepresented in all the studied sequences. Another dinucleotide, CpG, that is also generally underrepresented in the human genome, was randomly presented (≥0.78 and≤1.23) in ABCA7 and LRP1 sequences but overrepresented in APOE (≥1.23).
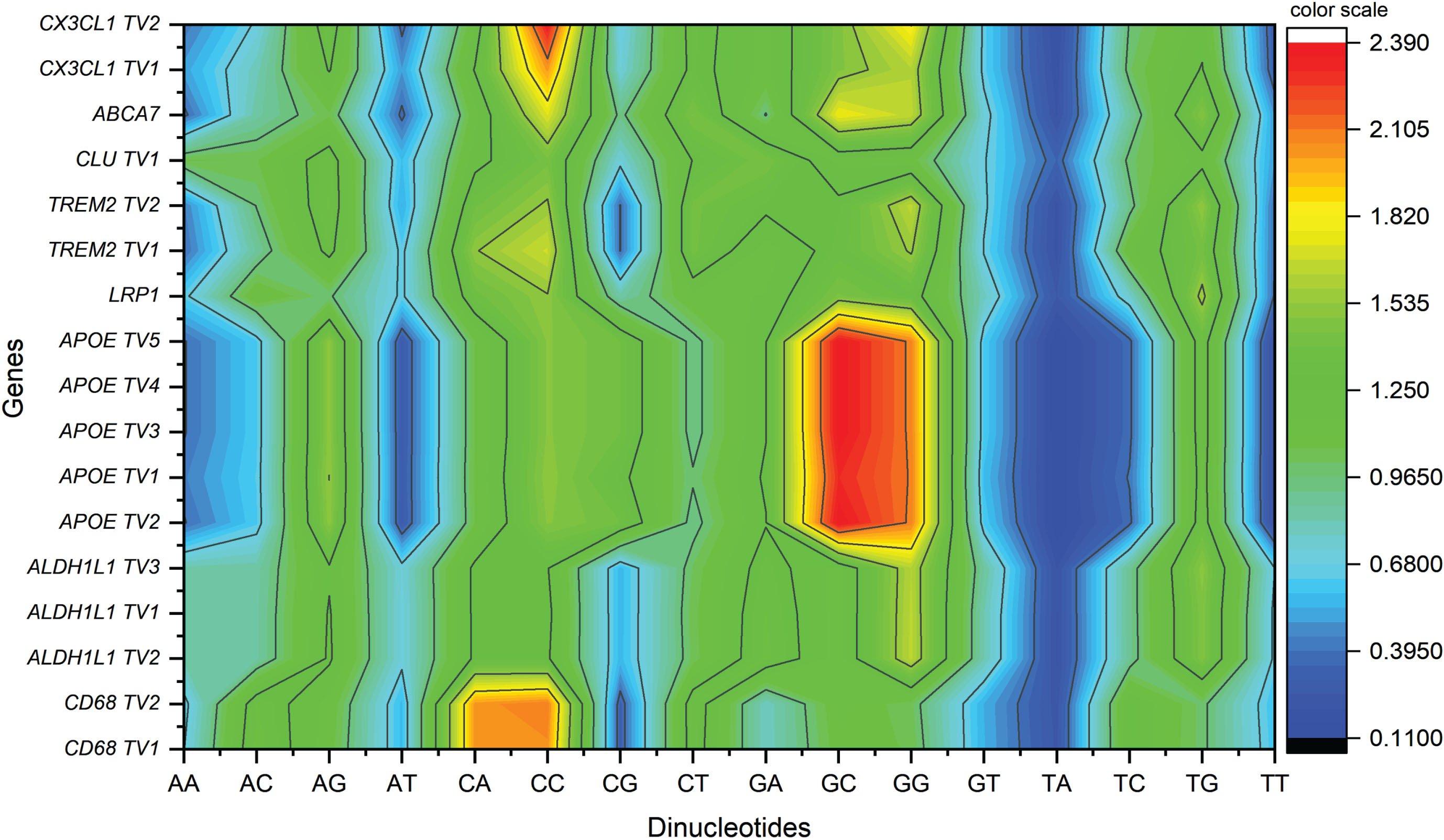
Countour plot for dinucleotde odds ratio.
Presence of bias at codon junctions
Dinucleotide TpA is disfavored at codon position p3-1 despite the fact that at this frame, TpA has no coding function [50, 51]. In the currently envisaged 17 transcripts, out of 16 codons encompassing TpA or CpG at the p3-1 junction, CpG presented a highly negative context (residual value< -75.0) for 12 codons. Compared to CpG, TpA dinucleotide at p3-1 junction had low negative context as demonstrated by a residual value less than -75 for only one amino acid, Asp, while it is least (more than -25) for Arg and Leu. The results suggested that CpG is more highly disfavored at junctions than TpA (Fig. 2).
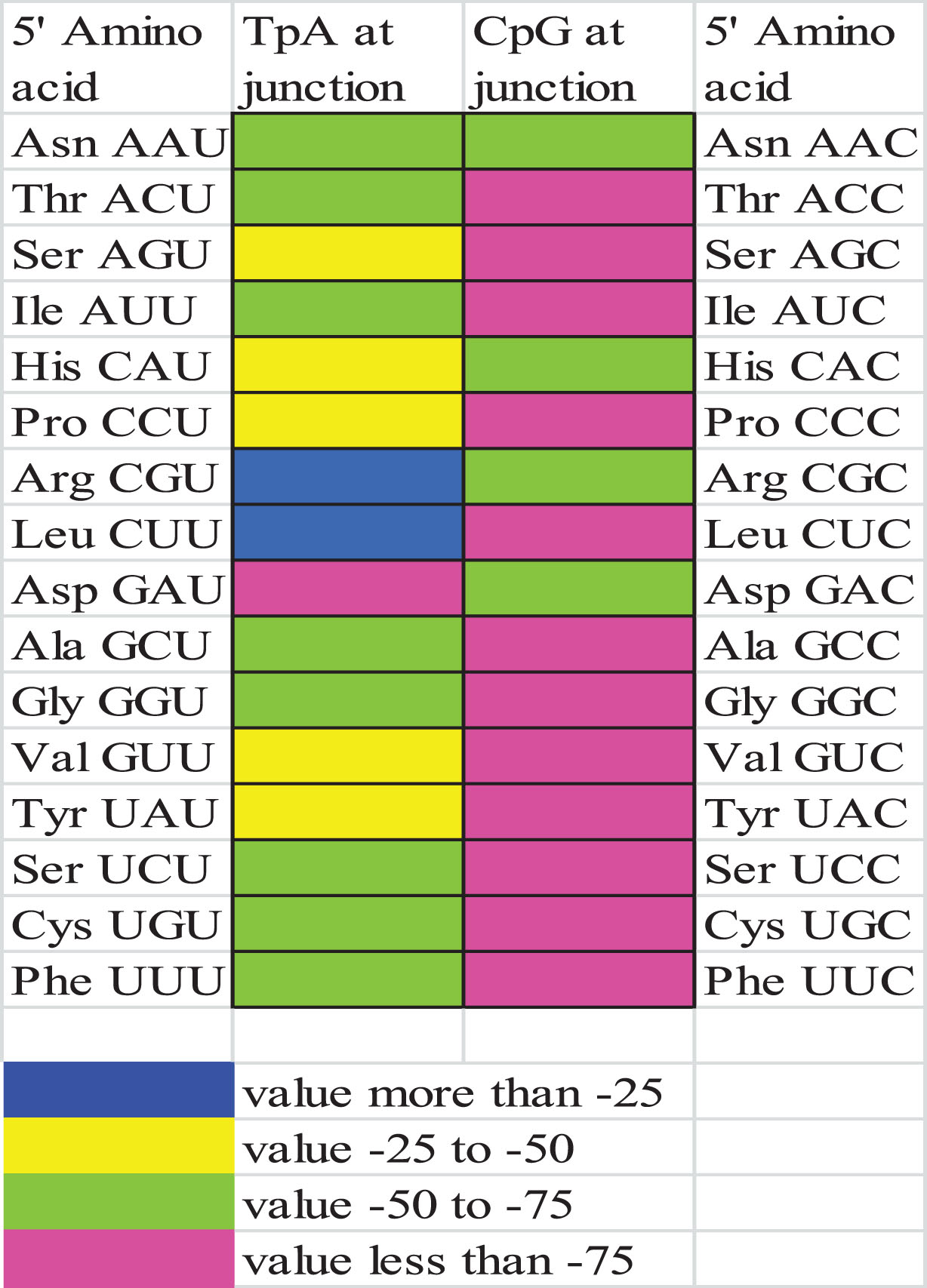
Color map for odds ratio of dinucleotides present at codon junctions.
RSCU analysis revealed that G/C ending codons were preferred
Codon usage is generally dependent on the genome composition [52]. Previous data has shown that the AT-rich genome prefers A/T ending codons, while the GC-rich genome shows G/C ending codons. RSCU values above 1.6 are said to be over-represented codons [53]. In the present study, we also investigated whether the same is true for our sequences. We aligned all the RSCU values of codons according to A/T or G/C ending codon, constructed a heat map based on the RSCU value, and found that our sequences are also GC-rich (Fig. 3). APOE transcripts showed even higher RSCU values suggestive of highly overrepresented G/C ending codons. Codons CAA, TTA, CTA, GTA, and CGT were underrepresented in all the sequences (RSCU values below 0.6), and all were A/T ending except for CGT. The RSCU value for CTG was the highest, ranging between 4.53 and 2.4. Codons TTG and TCG were the only exceptions that were G ending and showed underrepresentation in all the sequences (RSCU < 0.6). Codons CGA, TTA, GTA, CTT, TGT, TAT, ATT, AAT, CAT, AGT were not used in the APOE gene transcripts. Table 2 presents the RSCU values of 59 codons of 17 transcripts.

Heatmap based on RSCU using correlation distance and average linkage between gene transcripts and codons. Here four codons TTT, TTC, TTA, and TTG clustered separately, since they presented their RSCU values in very narrow range.
RSCU value of sequences envisaged. Overrepresented codons are given in bold. G/C ending codons were overrepresented with high RSCU values
The results of correlation analysis between codon bias (SCUO) and different protein properties
***p < 0.001, **p < 0.01, *p < 0.5; NS, non significant.
SCUO correlation with nucleotide composition at different codon position
SCUO was positively correlated with the % GC composition at the first and third codon position (SCUO with % GC1, r = 0.852, p < 0.0001; SCUO with % GC3, r = 0.956, p < 0.0001) and negative but insignificant correlation with % GC2 content. The correlation between codon bias and composition is suggestive of the dominant role of mutational forces rather than selection forces in shaping codon usage in any organism [54]. It has been proposed that shorter proteins are favored over longer proteins (both having similar functions) by selection forces in order to reduce energy expenditure [55], and codon bias and gene length are positively correlated in the case of Drosophila [56]. However, in the present study, no such correlation between the codon bias and gene length has been observed; our results are supported by the results of Yang et al., 2014 who found similar results [57].
Protein properties are correlated to the codon bias
Protein properties like GRAVY, hydrophilicity, and aromaticity have shown a correlation with the codon [44]. We extended the range of protein properties to be envisaged and performed a correlation analysis between nine protein properties and SCUO. Protein properties like GRAVY, hydrophilicity, and aromaticity have shown a correlation with the codon bias [44]. We extended the range of protein properties to be envisaged and performed a correlation analysis between nine protein properties and SCUO. We found a significant negative correlation between codon bias and acidic, basic, and neutral amino acid frequency, hydropathicity (GRAVY), and aromaticity (AROMA).
Dominance of selection force on codon usage
A neutrality plot suggests the balance between two major evolutionary forces i.e. selection and mutation. Based on the neutrality plot, it is clear that 21.47% mutation force, while 78.53% selection force shapes codon usage in genes, modulated post-anesthesia, and involved in AD (Fig. 4). There is a significant positive correlation between GC3 and GC12 that suggests that mutational bias is present at all codon positions [58], which is valid for our study also. The present study had a positive correlation between GC3 and GC12 (r = 0.513, p < 0.05).
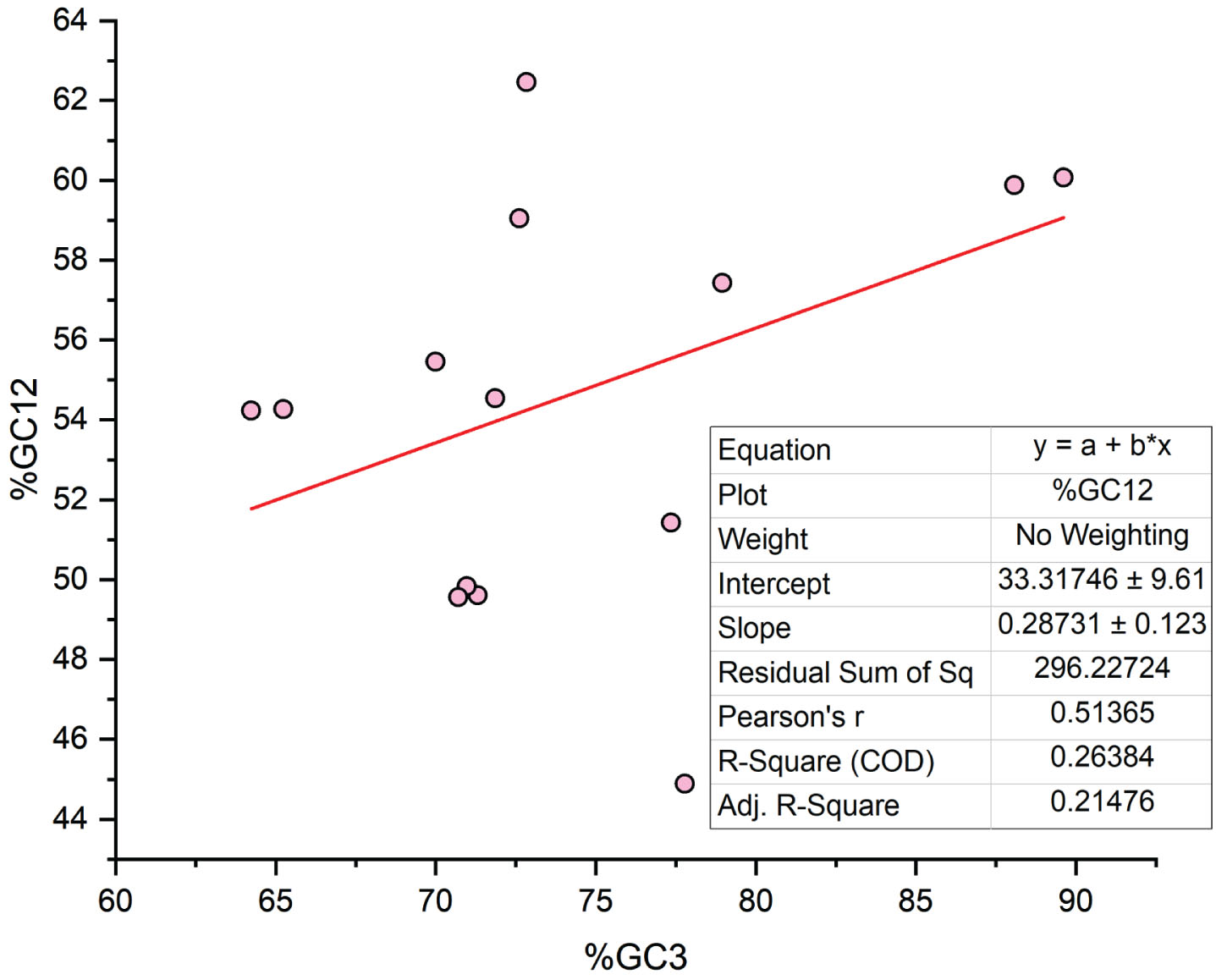
Neutrality plot analysis by regressing between % GC3 and % GC12 showing the equilibrium between selection and mutational forces.
T nucleotide is preferred over A and G and C nucleotides are used equally at third codon position
The parity plot reveals the bias between the AT and GC bias at the third codon position. According to Chargaff’s rule, the number of C residues in a DNA strand will equal G residues, and similarly, T residues will be equal to A residues [59]. However, practically, the situation deviates from Chargaff’s rule, and selection and mutational forces lead to deviation in the rule [60]. In the present study, the AT bias was 0.48±0.09 while the GC bias was 0.50±0.09. If the PR2 value is above 0.5, it suggests that purine is preferred over pyridine [61]. In the present study, AT was biased less than 0.5, and thus, T will be preferred over A. In the case of GC bias, the PR2 value is 0.5 and thus indicates that both the G and C are used equally at the third codon position (Fig. 5).
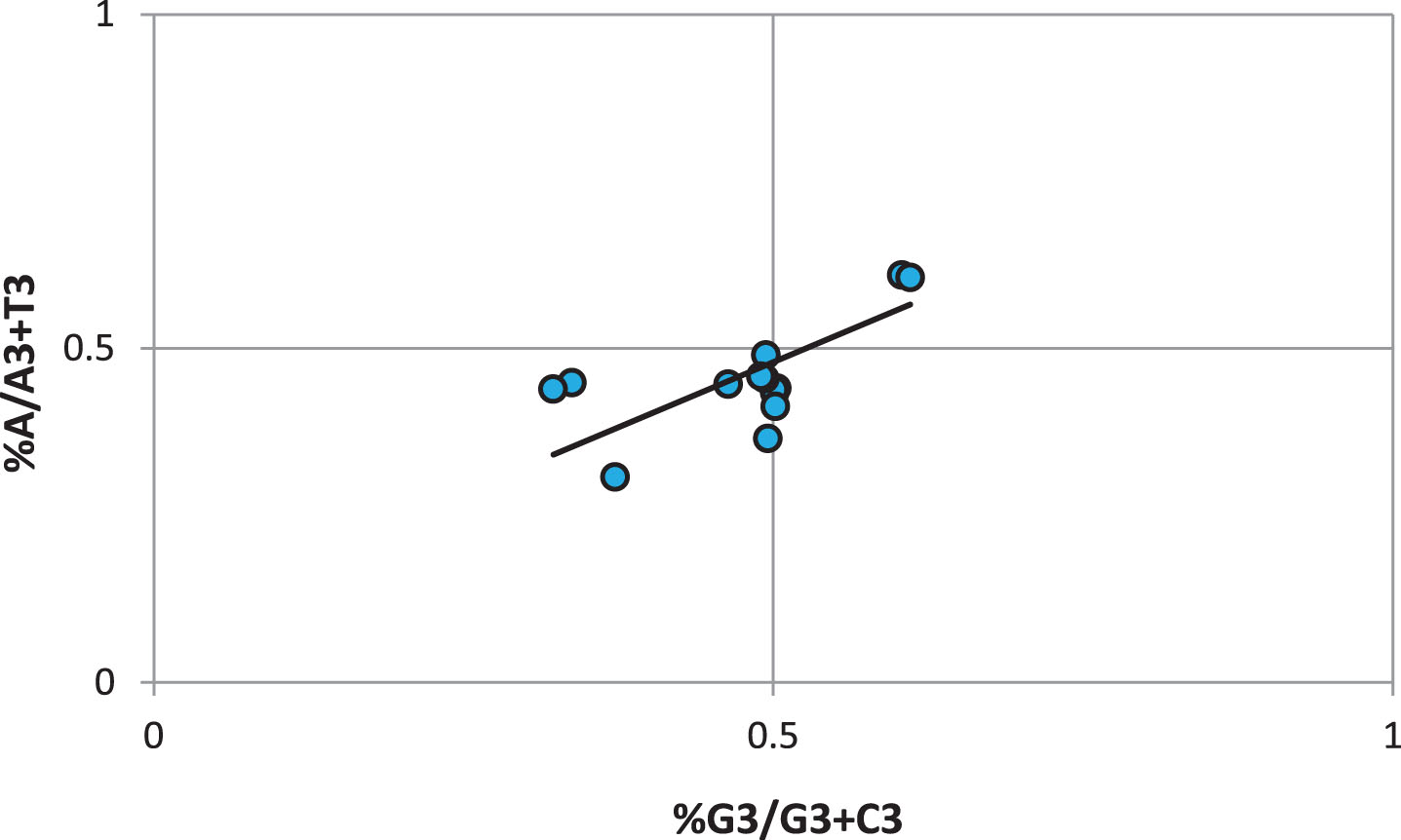
Parity plot exhibiting the AT and GC bias. Here, the X-axis represents the GC bias while the Y-axis represents the AT bias at the third codon position.
Nucleotide composition of cytosine showed unique feature
The percent composition of all four nucleotides had a statistically strong positive correlation with the length of the genes (p < 0.0001 for all nucleotides). However, only C nucleotide at the third codon position positively correlated with the transcript length, while other compositional parameters do not have (r = 0.670, p < 0.01). With that codon bias showed significant positive (% G, % G3, % GC, and % GC3) or negative correlation with other nucleotides compositions (% A, % A3, % T, % T3), but does not correlate with either overall % C or % C at the third codon position (% C3). Codon bias has been seen to be affected by nucleotide composition [62]. In the present study, overall composition and composition at the third codon position (% A, % A3, % T, % T3, % G, % G3) were correlated with codon bias in a statistically significant manner (p < 0.001, except for % A where it is p < 0.05). The only exception was nucleotide C, which has a correlation neither at overall composition nor at the third codon position with codon usage.
Highly occurring codon pairs have both 5’ and 3’ codon ending with C/G
Codon bias is a well-recognized phenomenon, and like the codon bias, there is a bias present in the codon pair, also called codon pair bias or codon context. It is sequential, unique to the genome, and a direct consequence of dinucleotide bias [63]. Both positive and negative contexts are visible in the studied transcripts. The matrix plot for codon context is given in Fig. 6.
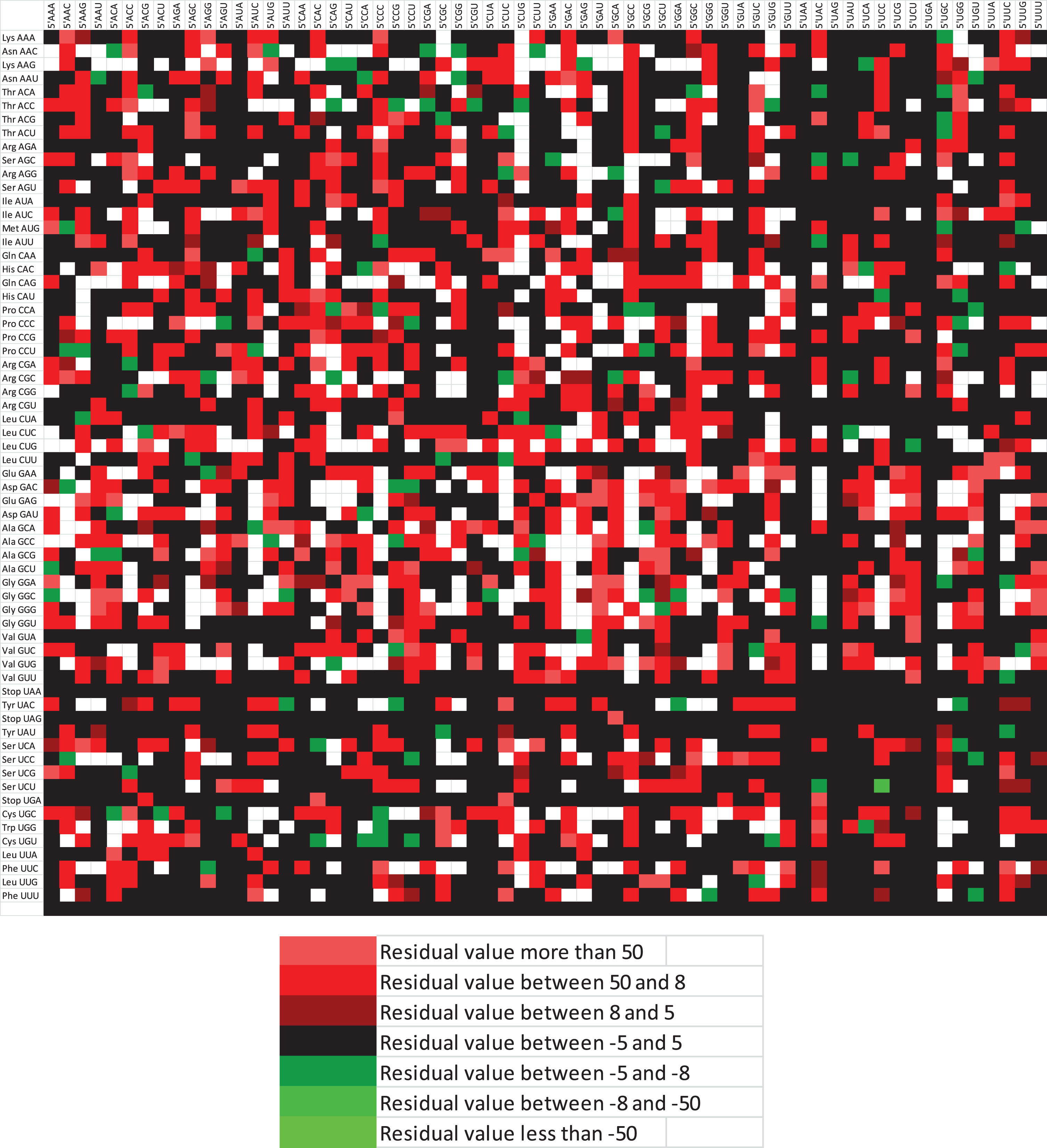
Matrix plot for residual values for codon context. Grey boxes are showing no context.
Of the total possible codon pairs, 1,740 were absent, and 308 codon pairs occurred only once in 17 transcripts. In the currently envisaged 17 transcripts, we found that all the highly preferred codon pairs (top 20 codon pairs) have both the 5’ and 3’ codons ending with G/C. Out of 20 codon pairs, seven codon pairs have 5’ codon Leu, while in the same number, it is present as 3’ codon also, signifying the role of Leu in the codon context (Table 4). Here, we observe an interesting phenomenon in an LRP1 transcript: stretches of 7 and 5 Leu residues (Fig. 7). In the ABCA7 transcript, a stretch of 7 residues was observed. In 17 transcripts, Asn, Glu, His, Met, Trp, and Tyr amino acids were present only as singlets or doublets but not as a stretch of more than two amino acids.
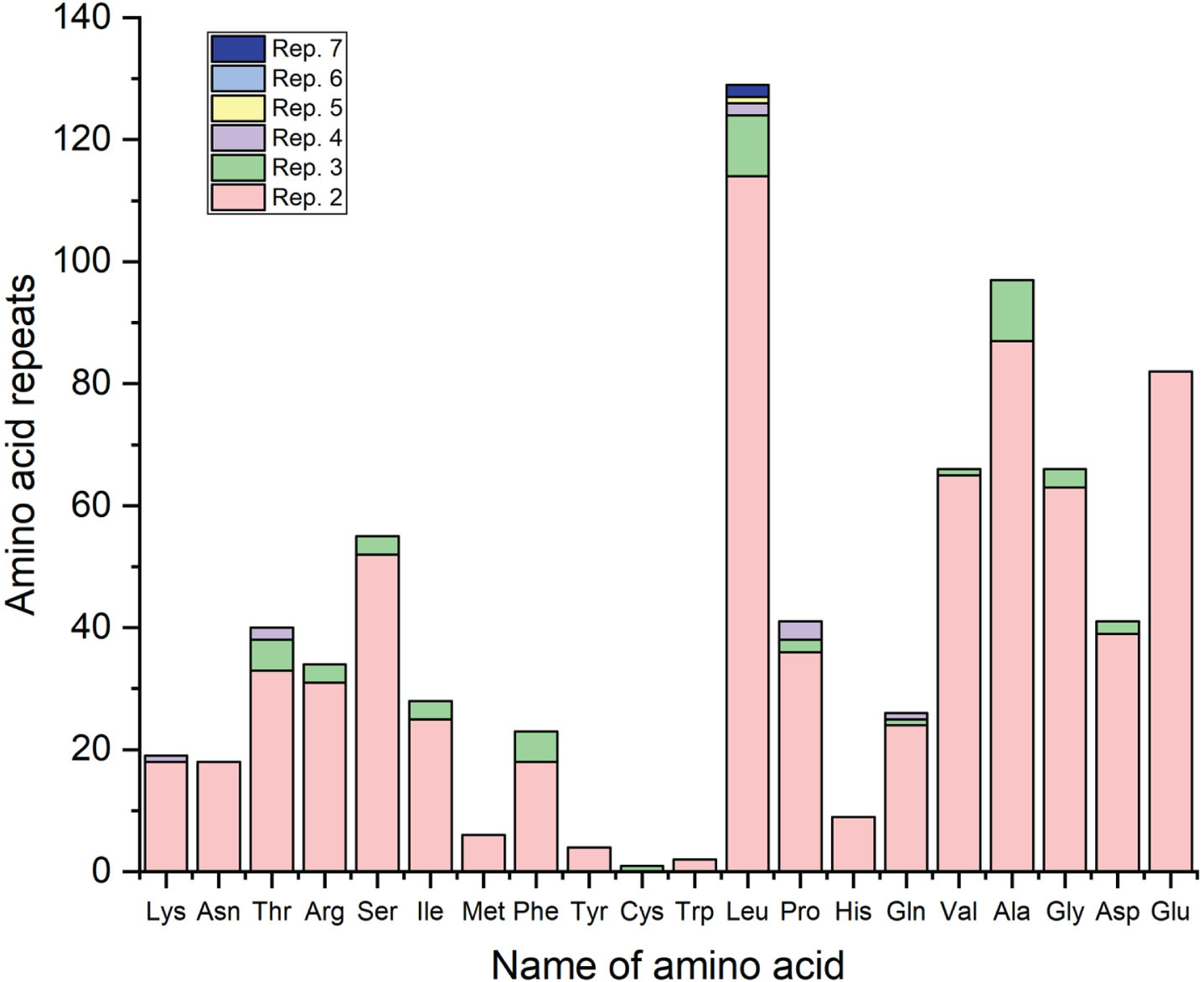
Number of times amino acid repeats were found in the envisaged 17 sequences. Leucine showed the highest number of Leucine doublets, while Cysteine showed the least, followed by Tryptophan.
Top 20 codon pairs in 17 studied transcripts with the number of occurrences
PCA analysis revealed maximum impact of codon AGC
PCA analysis based on RSCU values was carried out for 17 transcripts. Principle components 1 and 2 explained 64.59% and 11.675% variances, respectively. Overall, 90.3% variation was attributed to the first four axes. The analysis showed that the data points were scattered, suggesting that mutational force plays a role in shaping codon bias. Most of the data points were scattered far from the axis (Fig. 8A) and thus suggestive of much bias in codon usage [52]. Figure 8B is a biplot where the length of the arrow indicates the codon’s effect on codon usage. The lengthiest arrow for a codon shows the maximum impact on codon usage. In the present study, AGC, followed by CGC, poses the maximum effect on codon usage.
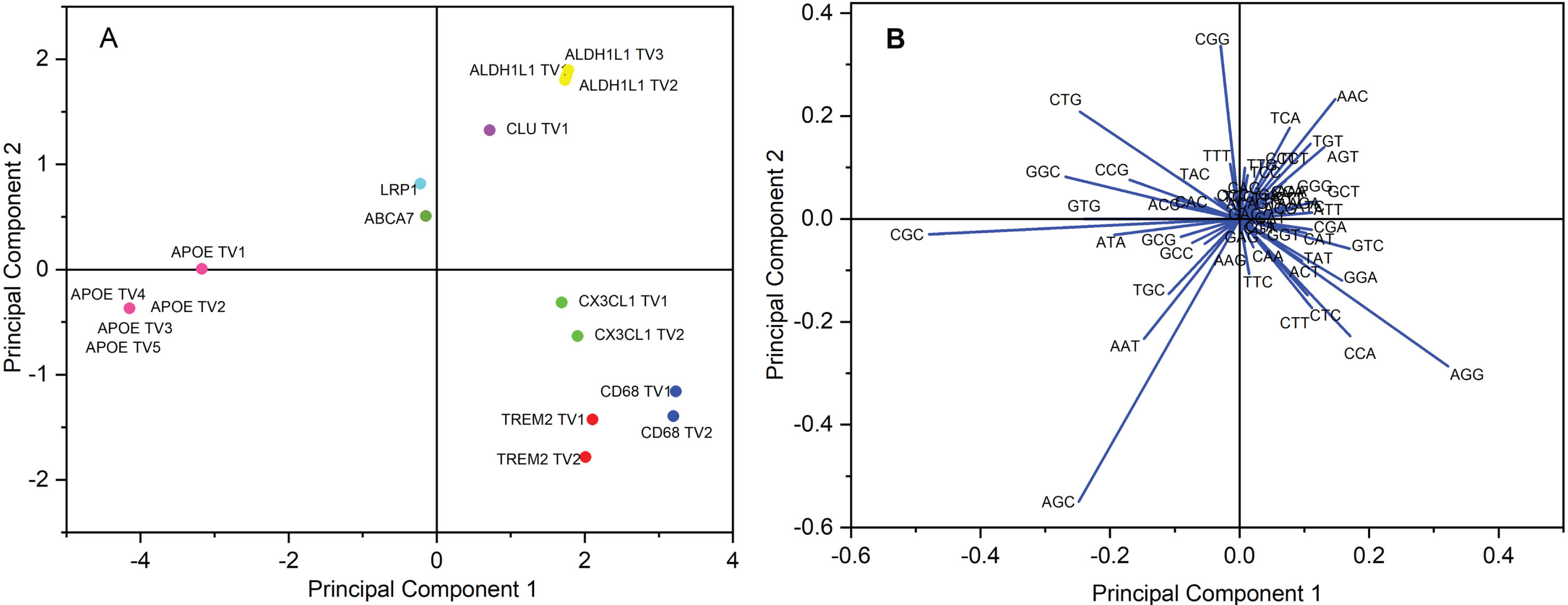
A) PCA analysis of 17 transcripts based on RSCU values. Data points scattered across the axis indicate that the RSCU values of each of the coding sequences are different from each other. However, transcripts of ALDH1L1 and APOE transcripts showed a similar codon usage pattern. Data points near the axes showed low codon bias. B) Biplot analysis for envisaged transcripts revealed a maximum effect of codon AGC on codon usage.
DISCUSSION
Nucleotide composition influences several features of the sequences, including codon usage [22], protein properties [23], and gene expression [24]. The nucleotide sequences we envisaged in the present study were GC rich, with overall slight abundance of C nucleotide over G, while at the third codon position, percent composition of both the C and G were almost equal. Since composition influences codon bias, compositional parameters are expected to be correlated with the SCUO. In the present study, we observed a unique molecular pattern that % C or % C3 that does not correlate with the SCUO, while other compositional parameters had. Nucleotide C composition demonstrated a statistically strong correlation with the gene length. Our results partially concord with the results of Hussain and colleagues [64]; protein length was negatively associated with % C. Such molecular pattern demonstrated by nucleotide C can be considered a unique feature of sequences triggers post-anesthesia, resulting in the initiation of AD associated pathology.
Nucleotide composition influences both the dinucleotide [65] and trinucleotide (codon) occurrence [22]. Being the sequence G/C rich, we expect G/C containing dinucleotides to be overrepresented, and the same was found faithful in our study. The dinucleotide CpC is overrepresented in all the genes (odds ratio (≥1.23)which can be explained based on the experiment of Khandia et al. (2022) [66] where overrepresentation of CpC was found in all the envisaged 31 transcripts and is suggested to link with the requirement of rapid demethylation machinery to fine-tune the Bim a proapoptotic gene involved in gene expression. GpG is overrepresented in all genes but in CD68 and CLU. CpG codon was underrepresented in CD68 and ALDH1L1, TREM2, CLU, and CX3CL1 transcripts, and it goes with the common feature of the human genome where CpG content is relatively low compared to the GC content present in the genome [67]. The possible reason for the low abundance is the negative selection of CpG dinucleotide. The CpGdinucleotides tend to mutate 42 times higher than any random mutation [68], possibly owing to the predisposal of CpG to deamination of 5-methylcytosine at CpG that lead to conversion of C to T and CpG is converted to TpG [69]. TpA dinucleotide is underrepresented in all the sequences, and its underrepresentation could be explained based on both the compositional forces [70] and selection forces [52]. Since TpA is an integral part of two stop codons, TAA and TAG mutation might lead to the formation of a stop codon between the sequence and premature termination of the protein. Apart from that TpA sequence-containing sequences are prone to degradation by cellular RNases, therefore selection forces tend to avoid the codons having TpA [71]. In the present study, the underrepresentation of TpA in all the sequences is attributed to compositional forces where both the A and T nucleotides are present less than C and G nucleotides. Mutation and selection are the two major evolutionary forces. Neutrality analysis reveals about the equilibrium between them. In the present study, selection forces were attributed majorly (78.53%) compared to mutational forces (21.47%) in shaping codon usage. The overall analysis suggests the presence of selection, mutation, and compositional forces in shaping codon usage in genes modulated post-anesthesia. In a previous study by Khandia et al. (2022) [28], they reported a correlation of length with codon bias, and contrary to their result in the present study, no correlation between codon bias and gene length was observed.
For the envisaged transcripts here, it is noteworthy that both the TpA and CpG had negative contexts at p3-1 positions, and it is different from the fact observed by Gurjar et al. (2023) [51], who demonstrated that presence of different contexts for different genes. For example, in the E gene of SARS-CoV-2 virus, only positive, while in the S gene, only negative context was present in the E gene for CpG dinucleotide. The results are suggestive of the fact that context is gene-specific and might be the result of selection force [50].
Nucleotide composition influences codon bias. Previous studies have demonstrated that A/T-rich genomes represent an overrepresentation of A/T-ending codons and vice versa. Underrepresentation of CAA, TTA, CTA, GTA, and CGT in all the sequences suggests that the underrepresented codons encompass TpA or CpG dinucleotide as an integral part of the codon except for the CAA. CTG has the highest RSCU values among all the coding sequences, and it concords with the results of Khandia et al. [39], who found an abundance of CTG codon in genes Common in Primary Immunodeficiencies and Cancer. C/G ending codons were overrepresented in the present study owing to compositional forces implicated in shaping codon usage. Not only were the codons biased towards G/C ending codons, but codon context was also affected by the same, and the top 20 highly occurred codon pairs had G/C ending codon (for both 5’ and 3’ codon).
Codon usage plays an important role in mRNA stability, which regulates mRNA stability through a common mRNA decay pathway. Furthermore, codon context (ATATTC at 3′ UTR motif) also destabilizes mRNA, signifying the effect of codon effect. The changes in codon context might result in the diseased condition that might arise due to the misfolding of protein, as demonstrated in the MDR1 gene where codon context variation leads to misfolded drug-transport MDR1 protein. It suggested that changes in codon context framework affects severely the elongation rate and protein folding efficiency and thus have a greater impact [61].
Leucine is known to stimulate the mammalian target of rapamycin (mTOR) that enhance protein expression and its overexpression has been known to attenuate inflammation-mediated cellular damage and dysfunction [72] and leucine-enriched proteins may modulate the inflammation [73]. The information gives us clues about the presence of a large number of repeats of leucine in envisaged sequences. Anesthesia exposure causes neuroinflammation [74]. Currently envisaged genes that are modulated post-anesthesia are leucine-rich, possibly owing to their role in protection from post-anesthetic neuroinflammation. Among all genes, ABCA7 and LRP1 have the most significant number of leucine repeats (seven). ABCA7 is expressed in neurons, astrocytes, microglia, endothelial cells of the blood-brain barrier, and brain pericytes and participates in the cerebral efflux of Aβ peptides and prevents their entry to the central nervous system. With a normal aging process, the levels of ABCA7 modestly decreased [75]. All pieces of evidence cumulatively point towards the protective role of ABCA7 against the inflammatory effect of anesthetics. If we observe the codon context, we find that in the codon context, leucine also showed a specific pattern. Out of the 20 most occurring codon pairs, seven codon pairs were initiated, and the same number ended withleucine.
Conclusion
AD decreases the quality of life of patients and their relatives. Studies conducted in Asia have shown the potential connection between anesthetic exposure and dementia. Aβ plaque and tau tangles are characteristic features of AD. Functional transcriptome-based analysis displayed differential expression of genes in the neurodegenerative pathways, and also post-anesthesia hyperphosphorylated tau aggregates have been observed in an experimental animal model. In the present study, we envisaged seventeen coding sequences that are differentially expressed post-anesthesia, and we found specific molecular features associated with these coding sequences. Both composition and selection forces contributed to codon usage, and the composition of nucleotide C was a specific manner. It was positively correlated with the length of genes and not with the codon bias.
The sequences were GC rich and CpG dinucleotide was overrepresented in all the five coding sequences of ABCA7 and LRP1 genes, suggestive of selection and compositional forces shaping dinucleotide bias. Though the CpG has abundance in the sequences, CpG had a highly negative context at the codon junction, suggestive of strong negative selection forces acting of p3-1 at the codon junction. GC ending codons and codon pairs having G/C in the 5’ or 3’ codons were favored, again underscoring the significance of compositional forces. Among the selection and mutation forces, 78.53% force was the selection force involved in shaping codon usage. Since the sequences are modulated post-anesthesia, which have neuroinflammatoryroles, the presence of high leucine repeats in envisaged sequences suggests the neuroinflammation quenching roles of these proteins and hence showed that molecular patterns present in coding sequences might explain the functional aspects also. The study elucidates the significance of compositional and selection forces and the molecular features envisaged in this study might open a new arena of functional analyses of genes modulated post-anesthesia.
Footnotes
ACKNOWLEDGMENTS
The authors acknowledge the support granted by respective institutes.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
Data will be made available upon request.