
Abstract
Background:
Rapidly progressive dementia (RPD), characterized by a rapid cognitive decline leading to dementia, comprises a diverse range of disorders. Despite advancements in diagnosis and treatment, research on RPD primarily focuses on Western populations.
Objective:
This study aims to explore the etiology and demographics of RPD in Chinese patients.
Methods:
We retrospectively analyzed 323 RPD inpatients at Huashan Hospital from May 2019 to March 2023. Data on sociodemographic factors, epidemiology, clinical presentation, and etiology were collected and analyzed.
Results:
The median onset age of RPD patients was 60.7 years. Two-thirds received a diagnosis within 6 months of symptom onset. Memory impairment was the most common initial symptom, followed by behavioral changes. Neurodegenerative diseases accounted for 47.4% of cases, with central nervous system inflammatory diseases at 30.96%. Autoimmune encephalitis was the leading cause (16.7%), followed by Alzheimer’s disease (16.1%), neurosyphilis (11.8%), and Creutzfeldt-Jakob disease (9.0%). Alzheimer’s disease, Creutzfeldt-Jakob disease, and frontotemporal dementia were the primary neurodegenerative causes, while autoimmune encephalitis, neurosyphilis, and vascular cognitive impairment were the main non-neurodegenerative causes.
Conclusions:
The etiology of RPD in Chinese patients is complex, with neurodegenerative and non-neurodegenerative diseases equally prevalent. Recognizing treatable conditions like autoimmune encephalitis and neurosyphilis requires careful consideration and differentiation.
Keywords
INTRODUCTION
Rapidly progressive dementia (RPD) is a definition of the cognitive disorders with fast progression contributing to clinical syndrome of dementia within a relatively short period (less than 1 or 2 years) [1, 2]. In contrast to most dementias that progress in more than 10 years of time, a majority of RPDs would progress rapidly and even become fatal in a short period [3]. RPD displays a wide array of etiologies, encompassing neurodegenerative disorders, such as Alzheimer’s disease (AD), frontotemporal dementia (FTD), and prion disease, as well as non-neurodegenerative disorders, such as cerebrovascular diseases, central nervous system (CNS) infections, metabolic and toxic diseases, and brain tumors [2, 4]. Creutzfeldt-Jakob disease (CJD), arising from the aberrant conformational changes in the prion protein, stands as a prevalent manifestation within the spectrum of RPDs [2, 5]. The conditions of CJD usually deteriorate rapidly and can lead to irreversible disability and even mortality in a short period of time [5]. Unlike CJD and other neurodegenerative disorders contributing to RDP, some etiologies of RPD are treatable and reversible, including autoimmune encephalitis [2]. Therefore, the urgent and early assessment and differential diagnosis of patients with RPD are necessary and important for the subsequent treatment and prognosis.
The etiological factors contributing to RPD are multifaceted, rendering the prompt diagnosis and treatment of RPD challenging. Previous investigations have mainly focused on studying the etiologies of RPD among patients initially suspected of having prion diseases [6 –8]. Furthermore, past literature has documented the spectrum and etiologies of RPD in Western populations, with CJD and infectious or immune-mediated disorders being the most commonly observed causes [9, 10]. There are few reports in China summarizing the etiology of RPD from single centers, and the sample sizes are relatively small [11], or lack sufficient pathological data to make a definitive diagnosis of the causes [12]. The characterization and underlying causes of RPD in the Chinese Han population are still largely unexplored and poorly understood.
Here, we conducted a three-year retrospective study of 323 patients with RPD admitted to the department of neurology at Fudan University Huashan Hospital. In this study, we conducted a comprehensive analysis of the clinical and epidemiological data of patients to elucidate the disease profiles of individuals with RPD in our research center. Our investigation advances beyond preceding research through the inclusion of PET or CSF analysis, yielding enhanced accuracy and depth in the etiological assessment of RPDs. Through rigorous data collection and subsequent analysis, we aimed to gain valuable insights into the characteristics and patterns of RPD.
MATERIALS AND METHODS
Subjects
This is a retrospective data collection study. Patients included in this study were all admitted to the Department of Neurology, Fudan University Huashan Hospital between May 2019 and March 2023. A total of 323 hospitalized patients with RPD were included in this study, and criteria for inclusion included: (1) patients with cognitive impairment (Mini-Mental State Examination (MMSE)≤22) and (2) the duration of the symptoms of cognitive impairment was less than one year.
All patients included in this study underwent physical and neurological examinations. In addition, all patients underwent routine blood tests (blood cell count, blood chemistry, C-reactive protein, hepatic, renal, and thyroid function, and syphilis serology) and cerebral magnetic resonance imaging including T1, T2, fluid-attenuated inversion-recovery sequences, susceptibility weighted imaging sequences. For patients who required a diagnosis and had no contraindications, lumbar puncture or positron emission tomography/computed tomography (PET/CT) was performed. The cerebrospinal fluid (CSF) obtained was tested for cytology, biochemistry, syphilis, and antibodies related to autoimmune encephalitis. Participants were recruited and provided written informed consent, which was approved by the institutional review board in the study.
Diagnosis
The diagnosis criteria for the etiology of RPD can be found in Supplementary Table 1. If an individual has a positive amyloid PET scan or positive amyloid in cerebrospinal fluid, they are considered to have biomarker-defined AD. In our classification, we not only categorized the type of disease, but also distinguished between neurodegenerative causes of RPD (e.g., AD, and rapidly progressive dementia with Lewy bodies) and non-neurodegenerative causes of RPD (e.g., autoimmune encephalitis, infectious, toxic, and metabolic causes).
Statistical analysis
Statistical analysis was carried out using SPSS 21.0 software (IBM Corporation, Armonk, NY, USA). T-test was used to compare the differences in terms of age, time of onset, baseline MMSE and Montreal Cognitive Assessment (MoCA) scores between neurodegenerative and non-neurodegenerative groups. A significance level of p < 0.05 was used to determine statistical significance. Data visualization was conducted in R software (version 4.2.0).
RESULTS
Demographic characteristics and etiologies of RPD
The demographic characteristics of the 323 RPD patients, based on their etiologies, are presented in Table 1. Male subjects accounted for 53.9% of the sample, with a mean age of onset of 60.65±13.30 years and a mean duration of onset of 6.91±4.54 months. The average MMSE score was 13.19±6.97. Among the RPD cases, 128 (39.6%) had a course duration of less than 6 months. Memory loss was the most common primary symptom reported by RPD patients, accounting for 64.7% of the cases, followed by behavioral abnormalities (18.9%) and language dysfunction (11.5%).
Demographic and Clinical Data
Clinical symptoms and behavioral symptoms are the initial symptoms. MMSE, Mini-Mental State Examination; RPD, rapidly progressive dementia.
The etiologies of RPD are shown in Table 2. The most common etiology of RPD was neurodegenerative and vascular dementias, accounting for 153 cases (47.4%), This was followed by inflammatory CNS diseases, accounting for 100 cases (31.0%), and other cases, accounting for 41 cases (12.7%).
Characteristics of patients with rapidly progressive dementia in current study
Data presented as mean±standard deviation unless stated otherwise. RPD, rapidly progressive dementia; CNS: central nervous system. Other Neurodegenerative disorders: frontotemporal lobar degeneration, normal pressure hydrocephalus, motor neuron disease, spinocerebellar ataxia, Huntington’s disease, and neuronal intranuclear inclusion disease.
In the overall RPD patient population, the distribution of diseases showed that autoimmune encephalitis had the highest proportion, accounting for 54 cases (16.7%). AD had the second-highest proportion, with 52 cases (16.1%), followed by neurosyphilis with 38 cases (11.8%), and CJD with 29 cases (9.0%). Other neurodegenerative disorders include 17 cases of FTD, 7 cases of normal pressure hydrocephalus (NPH), as well as motor neuron disease (MND), spinocerebellar ataxia (SCA), Huntington’s disease (HD), and neuronal intranuclear inclusion disease (NIID). The Mimics of RPD include 9 cases of psychiatric disorders and 9 cases of other conditions. Among the other conditions are chronic subdural hematoma, symptomatic epilepsy, mitochondriopathies, and cerebral white matter malnutrition with uncertain causes.
The distribution differences of age and baseline MMSE or MoCA scores among different etiologies were calculated. It was found that there was no statistical difference in terms of age between neurodegenerative (62.18±10.00) and non-neurodegenerative groups (60.30±13.93; p = 0.324). In addition, there was no statistical difference in MMSE scores (13.33±5.11 versus 13.15±7.33, p = 0.856) or MoCA (7.43±5.27 versus 8.07±5.95, p = 0.447) scores between the two groups, either. As expected, there was a statistically significant difference in the duration from first symptom to dementia syndrome between neurodegenerative and non-neurodegenerative etiologies (10.48±2.99 months versus 6.09±4.44, p < 0.001).
The neurodegenerative and non-neurodegenerative causes of RPD
Overall, the proportion of neurodegenerative and non-neurodegenerative causes of RPD is about 48% (Fig. 1A). The various etiological factors within neurodegenerative and non-neurodegenerative categories of RPD are detailed in Fig. 1B and 1C. Of all patients, autoimmune encephalitis was the most common cause, followed by neurosyphilis, then by CJD (Table 3). Then based on whether the course of the disease is less than 6 months, our subgroup analysis of RPD etiology found that AD was the most common cause in neurodegenerative disorders, which accounted for 16.10% of all RPD cases, followed by CJD (8.98%) and frontotemporal dementia (FTD, 5.26%). Among non-neurodegenerative conditions, autoimmune encephalitis was the most common cause, which accounted for 16.72% of all RPD cases, followed by neurosyphilis (11.76%) and vascular cognitive impairment (8.05%). In neurodegenerative causes of RPD 64.10% had a course of 6 months or more, while in non-neurodegenerative causes, 73.44% had a course of less than 6 months (Table 3). However, some exceptions should be noticed. For instance, over a half of CJD cases presented with RPD less than 6 months. Moreover, there were a larger proportion of RPD more than 6 months in patients with neurosyphilis.
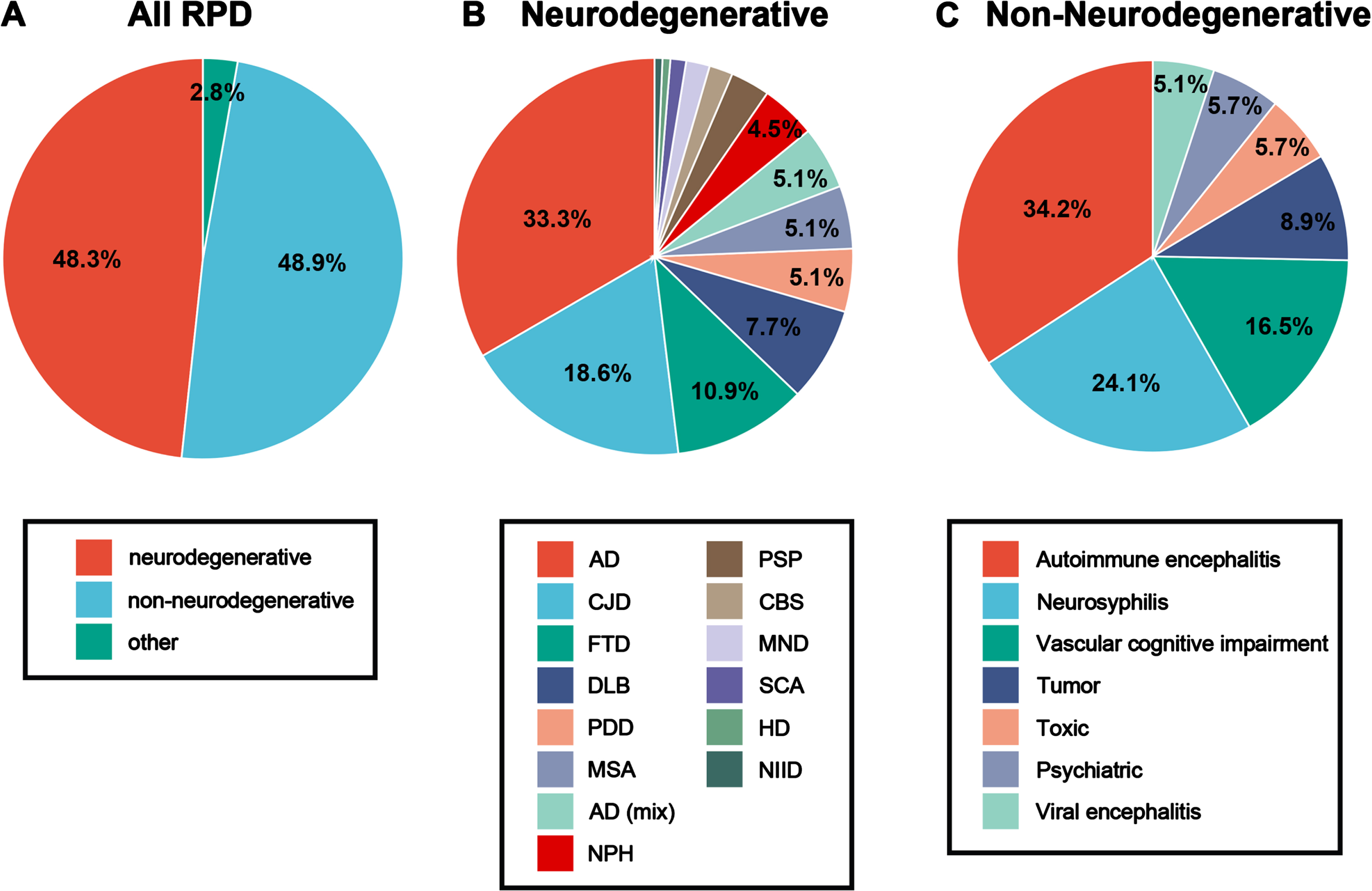
Spectrum and frequency of rapidly progressive dementia based on time course
RPD, rapidly progressive dementia; AD, Alzheimer’s disease; CBD, cortical basal ganglia degeneration; CBS, cortico-basal syndrome; FTD, frontotemporal lobar degeneration; CJD, Creutzfeldt–Jakob disease; PDD, Parkinson’s disease; DLB, dementia with Lewy body; MSA, multiple system atrophy; PSP, progressive supranuclear palsy; NPH, normal pressure hydrocephalus; MND, motor neuron disease; SCA, spinocerebellar ataxia; HD, Huntington’s disease; NIID, neuronal intranuclear inclusion disease. # Others: the diseases that are rare or cannot be classified according to the current state of research, including 2 chronic subdural hematoma, 2 symptomatic epilepsy, 3 mitochondrial disease, and 2 leukodystrophy with uncertain causes.
The etiologies of RPD with different courses of disease
For the cases with a course of the disease less than 6 months, the most observed etiology was autoimmune encephalitis with 30.47%, followed by CJD with 12.50% and neurosyphilis with 11.72% cases. For the cases with a course of the disease greater than 6 months, AD was the most observed etiology, with 51 (26.15%) cases, followed by neurosyphilis with 23 (11.79%) cases and FTD with 16 (8.21%) cases.
DISCUSSION
This study aimed to provide a comprehensive profile of the clinical and etiological characteristics of RPD within Chinese Han population cohort, which consisted of 323 individuals. Our findings revealed that autoimmune encephalitis, AD, neurosyphilis, and CJD were among the major causes of RPD in our memory center. A significant proportion of RPD cases in this cohort were attributed to non-neurodegenerative causes. This suggests that RPD may be treatable and reversible in certain conditions, emphasizing the importance of timely identification and diagnosis of the underlying etiology associated with RPD.
Dementia, which is one of the leading causes of disability and dependency in older individuals [13, 14], often has an insidious onset and a chronic progression, whereas RPD usually has a rapid progression, peaking in even weeks or months [1]. RPD is a vague definition and was initially considered one of the typical clinical manifestations and diagnostic criteria of CJD [1]. In recent years, there were increasing observational studies suggesting that in addition to CJD, a variety of neurological conditions or secondary conditions led to RPD, and some of the causes were treatable, like immune-mediated diseases, metabolic and toxic conditions, and other secondary neurological disorders [15, 16]. The incidence of RPD among dementia patients varied from 3.7–27% based on different definitions [17, 18]. Owing to the great number of dementia patients, RPD posed a heavy socioeconomic burden on public health care and was regarded as a major challenge for the diagnosis and treatment of dementias [14]. Comprehensive profiling and in-depth analysis of the etiologies behind RPD will yield critical insights essential for accurate differential diagnosis. Our analysis suggested that reversible diseases accounted for a large proportion of RPD causes. It is thus important for neurologists to identify the underlying treatable causes of RPD timely.
Supplementary Table 2 shows the distribution of RPD etiologies across different tertiary centers in multiple studies, and the observed frequencies are highly dependent on the study design. In our cohort, the most common cause of RPD was autoimmune encephalitis, followed by AD, neurosyphilis, and CJD. Immune-mediated encephalitis was one of the most important differential diagnoses for RPD [17 –19]. The typical symptoms of autoimmune encephalitis included early occurrence of altered consciousness and seizures [2]. Though immune-mediated encephalitis could be detected by certain antibodies, like the N-methyl-D-aspartate receptor (NMDAR) and leucine-rich glioma inactivated protein 1 (LGI1) [20], the diagnosis of immune-mediated encephalitis was challenging as the associated antibody could not be identified in some conditions [21]. Fortunately, once receiving appropriate treatment (i.e., corticosteroid and plasma exchange), the prognosis of immune-mediated encephalitis was usually better than other neurological disorders [22]. Our study showed that RPD due to autoimmune encephalitis tended to be younger and have a shorter time course of disease, which probably guide the differential diagnosis of RPD due to autoimmune encephalitis. In addition, CJD was the fourth common etiology for RPD in our cohort. Nevertheless, sporadic CJD was reported as the most common cause of RPD in another cohort at the University of California, San Francisco (UCSF) that included 178 cases in a period of six years [4, 23]. Our cohort was established within a diagnosis and treatment institution specializing in central nervous system diseases, with a primary focus on cognitive impairment as a complication, setting it apart from other referral hospital of certain diseases like CJD. Our study also indicated that non-neurodegenerative causes of RPD, including neurosyphilis and vascular cognitive impairment, contributed to a large proportion of RPD, suggesting that a lot of RPD patients were reversible with timely antisyphilitic treatment and controls of vascular risk factors, respectively. Another point worth noting is that leukodystrophy and mitochondrial disorders are also significant causes of RPD [24]. Early diagnosis of leukodystrophy offers patients potential treatment options, such as allogeneic hematopoietic stem cell transplantation and gene therapy, which hold promise [24 –26]. Overall, our study provided the memory diagnosis and treatment centers with reference for the differential diagnosis of RPD.
In our study, the most common neurodegenerative causes of RPD was AD, which was also the second most common cause of RPD. Consistent with our findings, Zhang et al. analyzed the major causes of RPD in the Chinese Han population and found that AD was the most common neurodegenerative cause [12]. Liu et al.’s findings echo our identification of neurodegenerative diseases and infectious as a leading cause of RPDs but note a higher incidence of toxic and metabolic conditions than we report [11]. The possible reasons for the difference among the studies were the differences in type of health-care system and ethnicities. Dementia due to neurodegenerative diseases, such as AD, FTD, dementia due to Lewy bodies, multiple system atrophy, or progressive supranuclear paralysis, was difficult to be found at the early stage and had a relatively slow progression of symptoms. Josephs et al. reported that AD, FTD, and DLB were all possible causes for RPD, particularly the patients with a disease duration beyond 12 months [27]. Likely, in another Western cohort of RPD patients, AD was reported to be one of the most common causes [19]. With the launch of anti-amyloid antibodies, such as lecanemab, new treatment options and opportunities for patients with AD have been provided [28, 29].
We also analyzed the etiology and spectrum of RPD in different courses of disease, which suggested that the most common disease of RPD with a duration of less than 6 months was autoimmune encephalitis, while AD was the most common etiology for RPD with a duration of more than 6 months. In addition, we found that neurosyphilis was one of the most common etiologies in RPD patients with a short or long course of disease. In China, the incidence of syphilis is increasing in recent years [30, 31]. However, the reported incidence of syphilis in China is dependent on case reporting, which may significantly underestimate the overall number of neurosyphilis. However, in our cohort, neurosyphilis accounted for a relatively higher proportion of RPD. The finding further highlighted the timely identification and diagnosis of RPD patients with short course and the subsequent etiological therapies.
The strength of this study was the relatively large number and complete clinical information of the RPD patients, but there were some limitations in our study. First, all RPD patients in the cohort came from the single center, which contributed to selection bias and an over-or under-representation of RPD patients. For instance, it was noted that a handful of RPD patients due to viral encephalitis were included in our study. Secondly, the observational design based on medical records made the clinical information limited, like the time of first symptoms and the onset.
Conclusions
In conclusion, our study reveals that RPDs constitute a heterogeneous group characterized by a broad spectrum of etiologies and manifestations. We found that both neurodegenerative and treatable non-neurodegenerative conditions frequently account for RPDs, with several non-neurodegenerative cases being reversible upon timely and effective intervention. Therefore, comprehensive assessments, encompassing diagnostic tests and imaging, are imperative for the prompt diagnosis and initiation of appropriate treatment strategies.
AUTHOR CONTRIBUTIONS
Qin Shi (Data curation; Formal analysis; Investigation; Methodology; Writing – original draft); Wei-Shi Liu (Conceptualization; Investigation; Methodology; Writing – original draft); Fang Liu (Writing – review & editing); Yi-Xuan Zeng (Data curation; Formal analysis); Shu-Fen Chen (Data curation; Formal analysis; Investigation; Writing – original draft); Ke-Liang Chen (Investigation; Methodology; Resources); Jin-Tai Yu (Funding acquisition; Resources; Writing – review & editing); Yu-Yuan Huang (Writing – original draft; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
Li Ren from Changzhou Geriatric Hospital is acknowledged for her help with acquisition of data.
FUNDING
This study was supported by grants from the Research Fund of Shanghai Municipal Health Commission (20234Y0307) and the National Natural Science Foundation of China (9184910220, 91849126, 81571245, and 81771148).
CONFLICT OF INTEREST
Jin-Tai Yu is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article.