
Abstract
Neurodegenerative disorders involve progressive dysfunction and loss of synapses and neurons and brain atrophy, slowly declining memories and cognitive skills, throughout a long process. Alzheimer’s disease (AD), the leading neurodegenerative disorder, suffers from a lack of effective therapeutic drugs. Decades of efforts targeting its pathologic hallmarks, amyloid plaques and neurofibrillary tangles, in clinical trials have produced therapeutics with marginal benefits that lack meaningful clinical improvements in cognition. Delivering meaningful clinical therapeutics to treat or prevent neurodegenerative disorders thus remains a great challenge to scientists and clinicians. Emerging evidence, however, suggests that dysfunction of various synaptogenic signaling pathways participates in the neurodegenerative progression, resulting in deterioration of operation/structure of the synaptic networks involved in cognition. These derailed endogenous signaling pathways and disease processes are potential pharmacological targets for the therapies. Therapeutics with meaningful clinical benefit in cognition may depend on the effectiveness of arresting and reversing the neurodegenerative process through these targets. In essence, promoting neuro-regeneration may represent the only option to recover degenerated synapses and neurons. These potential directions in clinical trials for AD therapeutics with meaningful clinical benefit in cognitive function are summarized and discussed.
Keywords
INTRODUCTION
Neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease, frontotemporal disorder, Huntington’s disease, and amyotrophic lateral sclerosis, have emerged as the most dreaded diseases due to a general lack of effective therapeutics. The number of patients suffering from AD, the most common neurodegenerative form of dementia, for instance, is currently numbered at more than 55 million worldwide. Neurodegenerative dementia due to AD in the elders is rising at an alarming pace and predicted to at least double by 2050 [1]. Dementia is a disorder of progressive loss of intellectual functions, such as attention, thought processing, reasoning, decision-making, language, and social behavior. The pathological hallmarks of AD have been defined as progressive formation of extracellular amyloidal senile plaques and intraneuronal tauopathy neurofibrillary tangles.
The high frequency of AD cases and urgent need to find effective therapeutics with meaningful clinical benefits in cognition have attracted many investigators and public-private entities to devote their efforts in AD drug development, currently numbering close to 200 at various phases of clinical trials [2]. “Cognition” is the mental process and action, including attention, learning and memory, thought and reasoning, processing language, and decision-making. These efforts have led to 7 approvals, including the recent three amyloid-β (Aβ) antibodies, aducanumab (now pulled from the market [3]), lecanemab, and donanemab [4–6], entering the market mainly through implementing biomarker-guided pipeline/approval. However, largely due to limited understanding of AD pathogenesis, pharmacological development of AD drugs that are capable of producing meaningful clinical therapeutic effects in cognition has not been very effective so far.
PATHOLOGIES OF AD
The lack of AD therapeutics with meaningful clinical benefit in cognition stems from a limited understanding of pathogenesis of this complicated disease. Several hypotheses about AD pathologies have evolved over the past hundred years, with each supported by investigational observations and discoveries, since the first AD case was diagnosed. The major ones include the cholinergic hypothesis, the glutamatergic hypothesis, the Aβ (Tauopathy) hypothesis, the ApoE cascade hypothesis, and the inflammatory (oxidant) hypothesis, from most of which limited success has been achieved with therapeutic drugs being approved for the clinic, and often with high risk of side-effects such as brain edema, hemorrhage, and/or atrophy.
At the center of AD pathology is the amyloid hypothesis [7], i.e., accumulation of neurotoxic Aβ leads to other AD pathological changes, including taupathy [8] and inflammation, and causes AD pathology progression of the disease, neurofibrillary tangles, cell loss, and vascular damage. Aβ is formed from metabolism of the amyloid precursor protein, through subsequent cleavages by β-secretase and γ-secretase. Metabolic cleavage by α-secretase, on the other hand, produces soluble sAβPPα, which promotes neurite outgrowth [9]. Dementia follows as a direct result of this Aβ deposition [10]. Autopsy studies of non-selected brains from 1- to 100-year-old, revealed that amyloidosis and tauopathy occurred younger when they occurred, in the locus coeruleus and lower brainstem, before puberty or in early young adulthood [11], than in in the trans-entorhinal regions. These were, however, potential AD cases had they lived longer. Catecholaminergic neurons and the brainstem may influence cognition. Nevertheless, there is no question whether amyloidosis and/or tauopathy impair functions of synapses and neurons [10, 12–14] as shown by numerous studies. Not clearly defined, however, is whether amyloidosis/tauopathy drives AD neurodegenerative progression from pathogenesis, and whether removal of amyloidosis/tauopathy predicts cognitive recovery and is essential to achieve any meaningful clinical benefits in cognitive functions for AD.
The roles of astrocytes in neuronal/synaptic communications cannot be ignored [15]. Astrocytes are involved in regulation of synaptic formation and stability, and their degeneration occurs very early in neurodegeneration [16]. Reactive microglia and astrocytes surround amyloid plaques and secrete pro-inflammatory cytokines, which are known to impair synaptic and neuronal structures and operation. However, long-term and population-wide usage of non-steroid anti-inflammatory drugs, including clinical trials, has not shown clinical benefits.
Synaptic/neuronal loss, often measured in the hippocampus and associative cortices, is a defining feature of neurodegeneration [17]. The hippocampus is among the earliest affected brain areas in AD [18]. Synapses are highly dynamic in size, shape, and numbers, and are closely linked to synaptic activity and strength [19, 20].
Importantly, synaptic operation and preserving target innervation require dynamic and lasting trophic signals. Neurotrophic factors include brain-derived neurotrophic factor (BDNF) [21], nerve growth factor (NGF), and glial cell-derived neurotrophic factor, affecting all types of neurons, such as cholinergic, glutamatergic, monoaminergic, GABAergic neurons [22] and non-neuronal cells as well [23]. BDNF is the most widely expressed neurotrophin in the brain of vertebrates, including the brainstem, basal forebrain cholinergic neurons, and other organs. It is especially abundant in the hippocampus and associative cortices, with critical functions from control of cell death to cognitive functions. Its receptor TrkB (tropomyosin receptor kinase B), which is primarily expressed in the brain, mainly mediates BDNF-induced signaling activities. Protein kinase Cɛ and BDNF signal pathway interact with α-secretase [24]. BDNF is positively associated with sustained high cognitive function late in life in AD [25]. Low expression levels of BDNF and its receptor TrkB lead to severe neuronal deficits, such as the decrease of dendrite length and spine densities in the hippocampal CA1 [26] and as well as synaptic and cognitive dysfunction, long before significant cell loss. Low levels of BDNF and TrkB have been found in the hippocampus and frontal cortices in AD brains [27–29], but not NGF and NT-3. In humans, loss of function BDNF/TrkB variants results in deficits in hippocampal synaptogenesis and cognition [30].
Understanding the interplay between amyloidosis/tauopathy, neuroinflammation, and neurotrophic activities in AD pathogenesis is interesting and as well as important. There is no lack of observations that amyloidosis/tauopathy and neuroinflammation [31] reduce neurotrophic activity in the brain. Neurotrophic deficits lead to synaptic neuronal breakdown in structures and operation and amyloidosis/tauopathy and neuroinflammation [32]. If amyloidosis/tauopathy and neuroinflammation, which is characterized by glial activation and overproduction of pro-inflammatory cytokines, plays a causative role in neurotrophic/synaptic deficits and AD pathogenesis, their blockade and removal/reduction should be expected to reverse neurotropic and signaling deficits in the brain and arrest AD progression and cognitive deterioration, much more than just the marginal cognitive improvement observed. On the other hand, if neurotrophic deficits are responsible for much of the brain amyloidosis/tauopathy and cognitive deficits, rescue of neurotrophic activity would be expected to reduce amyloidosis/tauopathy and arrest AD progression in cognition, unless amyloidosis/tauopathy does not directly mediate cognitive impairment due to neurotrophic deficits. In AD, gene expression of BDNF and TrkB was found to predict levels of amyloidosis and intracellular neurofibrillary tangles [33]. Attenuation of neurotrophic deficits has been suggested to abrogate amyloidosis and tauopathy [33]. Consistent with this potentially non-central role of amyloidosis/tauopathy in AD pathogenesis are also the evidence that the existence of significant amount of amyloidosis/tauopathy does not guarantee clinical consequence of dementia in the elders [34] and that prevention of the neuro-connections in the brains from breakdown, without removing Aβ and/or tauopathy, is sufficient to prevent dendritic spine loss, hippocampal neurodegeneration, and deficits in cognition [35]. In aged 3x Tg-AD mice, neural stem cell transplantation rescues cognition through BDNF-mediated robust enhancement of hippocampal synaptic density, without altering Aβ and tau pathology [36]. Loss-of-function studies show that knockdown of neural stem cell-derived BDNF fails to improve cognition and restore hippocampal synaptic density [36]. Viral delivery or overexpression of BDNF in a variety of AD models has also been found to improve cognition, independent of any effects on amyloid or tau neuropathology [37, 38].
CLINICAL CONSEQUENCES OF AD PROGRESSION
AD clinical consequences are characterized by a gradual deterioration of cognitive functions, functional independence, and behavioral changes. Daily routing activities are also affected eventually.
AD is usually divided into several stages according to the severity of the disease symptoms [39]. The most common are so-called “preclinical”, “mild”, “moderate”, and “severe/advance” stages, with some stages lasting many years. Mild cognitive impairment (MCI) represents an intermediate stage of cognitive decline with increased risk of AD progression. However, MCI may progress to normal cognitive stage over time [40].
Improvement of clinical consequences due to AD progression is the primary therapeutic goal in AD therapeutic development. Severe cognitive decline is a direct functional consequence of AD progression of degeneration [17], synaptic and neuronal dysfunction/loss (structure and operation) in cognitive networks such as the limbic and neocortical cortices.
Alzheimer therapeutic development has seen some compromises because of the unsure nature of the underlying pathogenesis responsible for the clinical consequences of AD. FDA’s guidance [41] for clinical drug trials in early AD for instance, has a 3-stage framework. Stage 1 exhibits abnormal biomarkers or detectable changes on sensitive measurements without cognitive complaints. Stage 2 is defined as subtle cognitive effects without functional deficits. Individuals beginning to experience difficulty with daily tasks are in Stage 3. The questions are thus whether the measured biomarker activities or biomarker-powered assessments reliably predict/reflect cognitive dysfunction and meaningful clinical benefits of cognitive functions. In two large phase III clinical trials, no evidence indicated biomarkers’ correlation with cognitive benefits [3]. Aducanumab and lecanemab were approved based on a reasonable likelihood that reduction of amyloid predicts clinical benefits [42]. Therapeutics from biomarker-powered clinical trials might be “problematic” in the future as we understand more of AD pathology, no matter how “reasonable” they appear at the time.
In addition to the AD hallmarks, amyloidosis and tauopathy, together with the AD pathology and progression exist four deficit profiles: 1) A deficit in expression/levels of neurotrophins and their receptors; 2) An inflammatory cytokine profile and cell infiltration [43]; 3) Lack/deficits of critical synaptic/network structures/operations; and 4) A reduced ability to maintain synaptic/network structures/operations. Cognitive tasks require timely operation of synaptic and neuronal communications [44]. The synapse, where functional connection and information transmission occur among neurons, is structurally and operationally dynamic and unstable. The dynamic control of spines and synapses represents the individual’s endogenous mechanisms/capacity in response to task demands [45, 46]. Synapse loss, an absolute predictor of dementia [47, 48], occur at early stages of AD [49], while cell death occurs at later stages. Synaptic loss, though relative, marks deficits in the capacity of the dynamic process in synaptogenesis and synaptic repair/remodeling according to task demands. The clinical phase of AD is initiated when the synaptic and neuronal reaction can no longer maintain the required network communications.
TARGETING TRIALS FOCUSING ON AD CLINICAL CONSEQUENCES
Amelioration of clinical consequences of neurodegeneration is essential in AD management. Cognitive impact, meaningful clinical benefit in cognition, represents one major outcome in clinical trials [50]. Meaningful clinical benefit in cognition of AD measures the rate of clinical progression due to AD, i.e., the impact of the underlying pathophysiology in AD at the group level. The most common measurements of cognitive function include the Mini-Mental State Examination (MMSE) [51], Alzheimer’s Disease Assessment Scale (ADAS)-Cog 12 [52], and the Alzheimer’s Disease Cooperative Study– Activities of Daily Living (ADCS-ADL) [53]. Meaningful clinical benefit in cognition describes an achievement with important value, such as a 25% defined as clinically meaningful decline [54], in cognitive functions change in those receiving treatment without severe adverse reactions versus placebo over the course of an 18-month clinical trial. Conducting randomized trials with cognitive benefits as endpoints is challenging for the need of large sample sizes and high costs. Alternatively, biomarker-powered clinical trials have also been conducted to reduce the sizes and cost. One issue is what underlying biological changes reliably reflect meaningful clinical effects in cognition [55].
The therapeutic development includes preventing/arresting progression of AD pathology, reversing neurodegeneration, and promoting neuroregeration. The former depends on how the AD pathogenesis is defined and understood. The latter, however, appears to have some promising outcomes through boosting endogenous capacity [56] of neuroregeneration. Cognitive decline is accelerated by the lack of cognitive stimulation and challenges [57]. Central to this stimulation is the activity of neurotrophic factors, which control survival and dynamics of neurons and synapses and as well as their functional operation [26, 58].
Non-pharmacologic approaches
Many non-pharmacological approaches have been suggested to improve cognitive functions and slow-down AD progression (Table 1). Long-term cognitive enrichment and healthy lifestyles are known to play important roles in cognitive resilience against neurodegenerative progression and cognitive decline [59–63]. Single-cell molecular atlas reveals that resilience to AD pathology is positively associated with several synaptic genes, including BDNF, and a better preservation of somatostatin inhibitory neurons in human prefrontal cortex [25]. Education, age, and gender have been found to be associated with different BDNF levels [64]. There is a wealth of data suggesting that diet and exercise increase BDNF levels in the brain and improve behavioral and cognitive functions [65, 66]. A randomized 24-week clinical trial revealed that cognitively enhanced Tai Ji Quan improved cognitive performance in older adults with MCI, better than standard Tai Ji Quan or stretching exercise [67]. The effects lasted more than 48 weeks. Intermittent fasting has shown improvement in cognitive function and contribute cognitive resilience through neurogenesis in the hippocampus [65]. A three-year trial of the MIND diet, cardiovascular Mediterranean and DASH diets, however, revealed no significant difference in cognitive function, brain health, and brain imaging from those who followed a mild caloric restriction diet [68]. The problem with improving synaptic/neuronal regeneration through life-style changes is its mild strength and enduring in duration, often insufficient to overcome neurodegeneration, especially for those who need it the most.
Some examples of potential anti-AD therapeutics
Synapses and neurons can also be stimulated physically. The integrity of generating activity synchrony underlies basic synaptic and brain health. Neurodegenerative pathologies dramatically impair cognition and synchronous synaptic/neuronal activity. Therapeutic potential of gamma stimulation (at 40 Hz), visual or auditory, noninvasive electric (such as transcranial current stimulation [69]) and magnetic stimulation, in treating AD has been preliminarily explored. Neurons, including neurons in the hippocampus, respond to 40 Hz sensory stimulation [70], generating a spread of rhythmic synaptic activity beyond sensory networks. Clinical trials show likely amelioration of neurodegeneration and dementia in AD [71–74]. Deep brain stimulation involves neurosurgical placement of electrodes within the brain to stimulate desired targets (18 clinical trials to treat cognitive disorders) [75]. Cell replacement therapy is another important option in AD therapy [76].
Pharmacological approaches
Enormous efforts and cost have been devoted to Alzheimer therapeutic development (Table 1). Early studies focused on enhancing cholinergic signaling since it was attenuated in early AD. Several decades of trials led to approval of 3 AD drugs through cholinergic and 1 through glutamatergic mechanisms. However, they all suffer controversial efficacy in clinical cognition [43]. The early drugs (donepezil, galantamine, rivastigmine, and memantine) only provide short-term symptomatic relief for the mild AD cases at best.
Amyloid formation in the brain has been believed by many to cause AD [77]. Its removal is expected to arrest the development of AD, thus an AD treatment. In AD trials, blocking formation of amyloidosis and hyperphosphorylated tau or enhance amyloid clearance have not seen their much success. Schenk and co-authors reported in 1999 that active Aβ immunization was able to reverse cognitive deficits in transgenic AD mice [10, 78]. Aβ clearance by active and passive immunotherapy has seen several failed clinical trials in early years [79–84]. Reducing Aβ production by targeting β-secretase and or γ-secretase in clinical trials was met with safety issues. Chronic (1–2 months) inhibition of β-secretase 1 was reported to have no impacts on steady-state dendritic spine density or morphology in the hippocampal CA1 or memory or cognitive flexibility in mice [85]. Scyllo-inositol and tramiprosate, small molecule Aβ binders, failed in clinical trials [86–88]. Several anti-Aβ antibodies have been found to effectively reduce amyloid deposits: Aβ, oligomers, fibrils, and amyloids, as detected using PET imaging [89]. But their cognitive benefits were none or rather limited [89–92]. Anti-amyloid and inhibition of tauopathy yield variable outcomes [93]. Trx0237, a tau aggregation blocker, did not show benefits in phase III trials [94] and so did tau vaccine [95]. The recent approvals of aducanumab (though now pulled from the market) lecanemab, and donanemab, mono-clonal anti-Aβ antibodies, are very encouraging, although their clinical benefits were minimal [96]. The use of anti-Aβ antibodies was, nevertheless, associated with significant safety issues, such as brain atrophy [97, 98], brain edema (17.3% with lecanemab), and hemorrhage (12.6% with lecanemab) [89, 100].
Promoting α-secretase expression/activity has been proposed to be one of the potential treatments for AD, with benefit effects reported [101, 102]. However, there is no direct in vivo evidence that α-secretase deficiency in the cognitive network is causative in AD pathogenesis.
Rapamycin, an immunosuppressive drug, is isolated from Streptomyces hygroscopicus. It inhibits the mechanistic target of rapamycin kinase (MTOR) and prolongs lifespan in all the animal species tested [103]. Rapamycin has thus been proposed for treatment of aging-related disorders and has been reported to reduce amyloidosis, tauopathy, and neuroinflammation and improve cognitive functions in a variety of AD models, suggesting that it may be an anti-AD drug candidate if used early enough [104, 105]. Two clinical trials (NCT04629495, NCT06022068) are being conducted to assess rapamycin for the treatment of mild AD.
Impacts of mimicking physical and cognitive enrichments on cognitive function/dysfunction may be achieved through pharmacological agents. Activation of PKCɛ increases PKCɛ expression/activity and activities of its downstream signaling pathway. Bryostatin-1, a relatively specific PKCɛ activator with BDNF-enhancing profile, including several pharmacological impacts such as anti-amyloidosis, anti-tauopathy, anti-inflammatory, antioxidant, was found at appropriate doses to successfully halt AD cognitive progression in a subset advanced AD patients with MMSE II 10–14 [106].
Potential of activating kinases for tissue protection and regeneration may lead to effective drugs against neurodegeneration (Fig. 1). PKCɛ activation increases endothelin converting enzyme activity and reduces amyloidosis [107]. PKCɛ may mediate protective effects of hypothermia [108] and ischemic preconditioning [109]. It has been shown that PKCɛ phosphorylates KATP+ channel in ischemic preconditioning to protect hippocampal CA1 neurons, an effect abolished by KATP+ blockers [109]. The brain contains 7-times more mitochondria KATP+ channel than heart [110] and the channel is known to couple energy metabolism to cellular electrical activity. Beneficial effects of kinase signaling, such as UCL-TRO-1938 (a small molecule activator of the PI3Kα isoform), include protection from ischemic injury [111–113] and enhance repair, neuroprotection/regeneration [111, 115]. Interestingly, taurine increases BDNF expression [116] and its deficiency functions as a driver of aging[117].
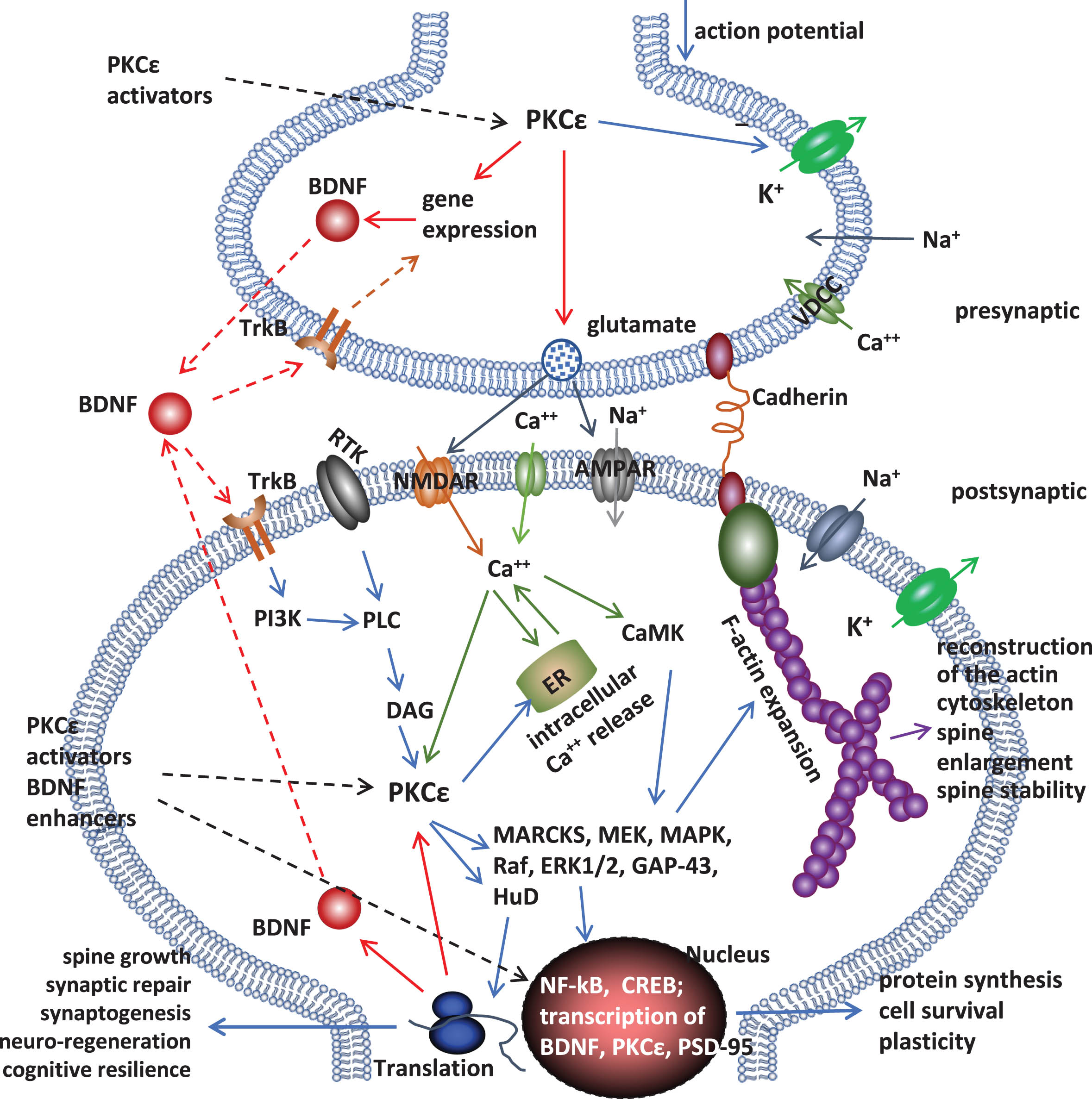
The synaptic PKCɛ– BDNF signaling pathway. Many signaling molecules are involved: such as mitogen-activated protein kinase (MAPK) kinase (MEK), MAPK, Raf, extracellular signal-regulated kinase (ERK) 1/2, and growth associated protein 43 (GAP-43), tyrosine kinase B (TrkB), phosphatidylinositol 3-kinase (PI3K), phospholipase C (PLC), and diacylglycerol (DAG). Through the signaling pathway, PKCɛ activators shift endogenous neuro-regeneration and synaptic/neuronal integrity (structures and operation) to support synaptic/cognitive functions.
FUTURE DIRECTIONS
Cognition, operation of the mind, is one of the most fearful abilities to lose. The gap in our understanding of critical pathological causes and their contribution to the clinical consequence of neurodegenerative disorders places a big hurdle for the success of AD therapeutic development.
Clinical trials of AD therapeutics are powered to detect outcomes of therapeutic interventions. The goal is to achieve at least meaningful clinical benefits in cognition. Although recent anti-Aβ clinical trials yielded some positive outcomes, their general no or limited clinical benefits after 18–24 months of treatment needs further clinical evaluation. Targeting APOEɛ4, the strongest genetic risk factor for late-onset AD, cascade has not yielded promising drug candidates so far. Some promising therapeutic outcomes through mainly neurotrophic pharmacology have been observed in therapeutic trials in advanced AD patients, arresting cognitive deterioration in advanced AD subjects with MMSE II of 10–14. It remains to be studied whether such treatment would provide longer clinical benefits in cognition and/or reverse the AD progression. Crucial for AD drug development is still our understanding of AD brain pathology.
AUTHOR CONTRIBUTION
Miao-Kun Sun (Funding acquisition; Writing – original draft; Writing – review & editing); Daniel Alkon (Supervision; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This study was supported by the National Institute on Aging of the National Institutes of Health (R44AG066366 to MKS) and sponsored by Synaptogenix, Inc.
CONFLICT OF INTEREST
Both authors are employed by Synaptogenix, Inc., developers of AD therapeutics.