
Abstract
Background:
Open-HART was an open-label extension of HART, a randomized, double-blind, placebo-controlled study of pridopidine in Huntington disease (HD). Previously, we reported safety and exploratory efficacy data after 36 months of treatment with pridopidine 45 mg twice daily. In the interim, emerging data suggests pridopidine may have neuroprotective effects mediated by sigma-1 receptor agonism.
Objective:
To report additional safety and exploratory efficacy data for continued open-label use of 45 mg BID pridopidine at 48 and 60 months.
Methods:
Patients in Open-HART were followed up to or greater than 60 months. Adverse events, concomitant medications, vital signs, laboratory values, and ECG data were monitored. Rates of decline in total functional capacity (TFC) and total motor score (TMS) over 60 months were evaluated in an exploratory analysis and compared between Open-HART and placebo recipients from the 2CARE trial. To account for missing data, sensitivity analyses were performed.
Results:
Of the original Open-HART baseline cohort (N = 118), 40 remained in the study at 48 months and 33 at 60 months. Pridopidine remained safe and well tolerated over the 60-month interval. TFC and TMS at 48 and 60 months remained stable, showing less decline at these timepoints compared to historical placebo controls from the 2CARE trial. TFC differences at 48 and 60 months observed remained nominally significant after sensitivity analysis.
Conclusion:
The 45 mg BID pridopidine dosage remained safe and tolerable over 60 months. Exploratory analyses show TFC and TMS stability at 48 and 60 months, in contrast to placebo historical controls from the 2CARE trial. Results are consistent with data reported from the recent Phase 2 PRIDE-HD trial showing less functional decline in the pridopidine 45 mg BID treated group at 52 weeks.
INTRODUCTION
Huntington disease (HD) is a fatal neurodegenerative disorder characterized by progressive motor, behavioral, and cognitive symptoms [1]. Significant unmet needs remain for symptomatic and disease-modifying therapies. Pridopidine, an investigational small molecule in HD, was initially postulated to influence motor symptoms of HD via modulation of dopamine receptors [2]. More recent data, including in-vitro binding assays and in-vivo PET imaging in rats, show pridopidine acts primarily via the sigma-1 receptor (S1R). Indeed, pridopidine demonstrates a binding affinity between 100 to 500-fold higher for S1R as compared to the dopamine D2 receptor [3, 4]. The S1R is an endoplasmic reticulum (ER) protein located mainly at the Mitochondria-Associated Membrane (MAM), where it regulates diverse cellular processes including calcium signaling, ion-channel modulation, and the ER stress response [5]. S1R activation is known to promote neuroprotection by stimulating neuronal survival, repair, and plasticity [6, 7].
Pridopidine demonstrates neuroprotective properties in several in-vivo and in-vitro HD models mediated by the S1R [6, 8]. A robust and dose-dependent neuroprotective effect against mutant huntingtin (mHTT)-induced cell death is observed after pridopidine administration in human HD iPSCs and in murine HD neurons, effects that are abolished by pharmacological inhibition of the S1R or S1R knock-out (KO) [9]. In the YAC128 model, pridopidine increases spine density in medium spiny neurons and rescues aberrant calcium signaling, both known features of HD. This effect is completely abolished in S1R KO neurons [6, 10]. Pridopidine reduces striatal aggregate size and upregulates brain-derived neurotrophic factor (BDNF) in the R6/2 HD murine model [11].
Pridopidine has been investigated in an HD ACR16 Randomized Trial (HART; NCT00724048), a 12-week, Phase IIb, randomized, placebo-controlled, dose-ranging, parallel-group study evaluating the effect of pridopidine at 10, 22.5, and 45 mg BID on motor symptoms in HD participants [12]. The effect of pridopidine on motor function was assessed after 12 weeks of treatment using the Unified Huntington’s Disease Rating Scale-modified Motor Score (UHDRS-mMS) as the primary endpoint. Other elements of the UHDRS, Total Motor Score (TMS) and Total Functional Capacity (TFC), were secondary endpoints. Although the HART study did not show a statistically significant difference between pridopidine treatment and placebo in the UHDRS-mMS, a nominally significant effect of pridopidine 45 mg BID relative to placebo on the UHDRS-TMS was observed (p = 0.039). Pridopidine at a dosage of 45 mg BID demonstrated a non-significant trend for improvement in TFC vs the placebo group (p = 0.09). A favorable safety and tolerability profile was observed at all doses, with no treatment-associated worsening of symptoms.
In a previous publication, we reported safety and exploratory efficacy data at 36 months for Open-HART, the open-label extension of the HART study [13]. The present analysis is a continuation of this effort, reporting additional safety and exploratory efficacy data at 48 and 60 months.
METHODS
Study population
The present analysis includes HD participants who successfully completed the HART study and continued into Open HART. Concomitant antidepressants and neuroleptics were allowed if treatment was stable for at least 6 weeks prior to study initiation and, as best as possible, remained unchanged throughout the study. Use of tetrabenazine or seizure threshold–lowering medications excluded subjects from participation. During the ongoing conduct of the open-label study, select medications known to prolong the QT interval were excluded.
Consenting process
An independent Ethics Committee or Institutional Review Board-approved informed consent was obtained from each patient prior to entering this study. Due to the possibility of progressive cognitive impairment impacting a patient’s ability to provide consent, each enrolled patient identified a research proxy. When appropriate, appointed research proxies were responsible for decisions regarding continued study participation.
Study design
Study structure remained similar from 36 months to 60 months, as described in the previous 36-month report [13]. Participant contact occurred every three months, alternating between the following types of visits:
Safety visit
An encounter, in-person or by phone, consisting of AE/concomitant medication review, vital signs, blood draws for electrolytes/creatinine clearance, ECG, and PBA-s. Some data could be collected locally by primary care physicians if optimal for the participant.
Clinic visit
An evaluation consisting of study consent, compliance assessment, review of concomitant medication and AEs, physical examination, ECG, vital signs and weight, blood draw for electrolytes and creatinine clearance, pregnancy test yearly when applicable, PBA-s, C-SSRS, and UHDRS.
Efficacy assessments
The UHDRS- TFC was evaluated as an exploratory endpoint [14]. This scale relies on clinician assessment of the patient’s ability to perform across five categories (capacity to work, finances, domestic chores, activities of daily living, level of care). Total scores range from 0 to 13, with higher scores indicating a greater capacity for independent function. TFC change was analyzed for the entire cohort, as well as separately in participants able to complete 60 months. TFC changes over 48 and 60 months were also compared to historical placebo participants from the Huntington Study Group–sponsored clinical trial 2CARE [15]. An early TFC group (baseline scores 9–13) was used for this comparison, as this was the cohort in 2CARE. Additional exploratory analyses were conducted comparing “early initiators” (patients randomized to any of the pridopidine arms in the original HART study who continued in Open-HART) to “late initiators” (patients randomized to the placebo arm in HART who received pridopidine only upon starting Open-HART).
The UHDRS-TMS was also used to evaluate efficacy in an exploratory manner. Each domain within the TMS was rated on a five-point scale from 0 (normal) to 4 (maximally abnormal) to generate a total score [14]. Similar analyses evaluating TMS change in 60-month completers and comparing TMS performance in Open HART to an early TFC cohort from 2CARE were also done.
Safety assessments
Safety visits included 12-lead ECG; assessment of vital signs; physical examination; blood draws to monitor calcium, potassium, and magnesium; and monitoring of AEs. Safety assessments were conducted at baseline and each subsequent in-person visit or telephone consultation. A treatment-emergent AE (TEAE) was defined as any AE occurring after the start of the study drug or the worsening of a preexisting AE or condition after the start of the study drug. As in previous studies of pridopidine, AEs of special interest included seizures/convulsions, cardiac conduction disturbances, and suicidal behaviors. The QT interval length was corrected using Fredericia’s method (QTcF) and was monitored, with prolongation defined as an interval of > 450 msec for both males and females or an increase of greater than 50 milliseconds above baseline or 60 milliseconds from any timepoint [16].
Concomitant medications were monitored at baseline and throughout the study.
Statistical analysis
Descriptive statistics of baseline characteristics as well as concomitant medication use and adverse events during the study were used to summarize the overall characteristics of the sample. Rates of adverse events were further delineated by year. Efficacy analyses for TMS, TFC, and the five individual functional capacity categories (capacity to work, finances, domestic chores, activities of daily living, and level of care) included mean and standard error (SE) scores as well as mean (SE) change from baseline at each yearly time interval for the entire cohort and the subset who completed the 60-months study.
Mean change from baseline for the TFC score, the five individual domains comprising the TFC, and TMS in the early HD cohort were compared to those randomized to placebo from the large, long–term HD study, 2CARE. Repeated-measures analysis of covariance models were used to compare estimates of treatment effect (pridopidine versus historical placebo) for each visit. Accounting for multiple comparisons, p-values < 0.01 were considered statistically significant.
In order to account for missing data after participant discontinued in both Open HART and 2CARE, a mixed model repeated measures (MMRM) with multiple imputations (100 datasets) approach was used to model missing data for subjects in Open HART and 2CARE using data available from other subjects within the study.
Exploratory analyses comparing “early initiator” and “late initiator” patients from the HART and Open-HART studies were performed as described above. These included summaries of the baseline characteristics, concomitant medication use and adverse events during the study, mean (SE) TMS and TFC scores and mean change (SE) from baseline at each yearly timepoint for the entire cohort and the subset who completed the 60-months study period.
RESULTS
118 participants who originally enrolled and successfully completed the HART study were re-enrolled into Open HART from 24 March 2011 to 07 December 2011. These participants were recruited from 22 sites in the United States and Canada. Figure 1 indicates participant disposition over the 60-month analysis period. Forty participants completed 48 months (33.9%) and 33 completed 60 months (30.0%).
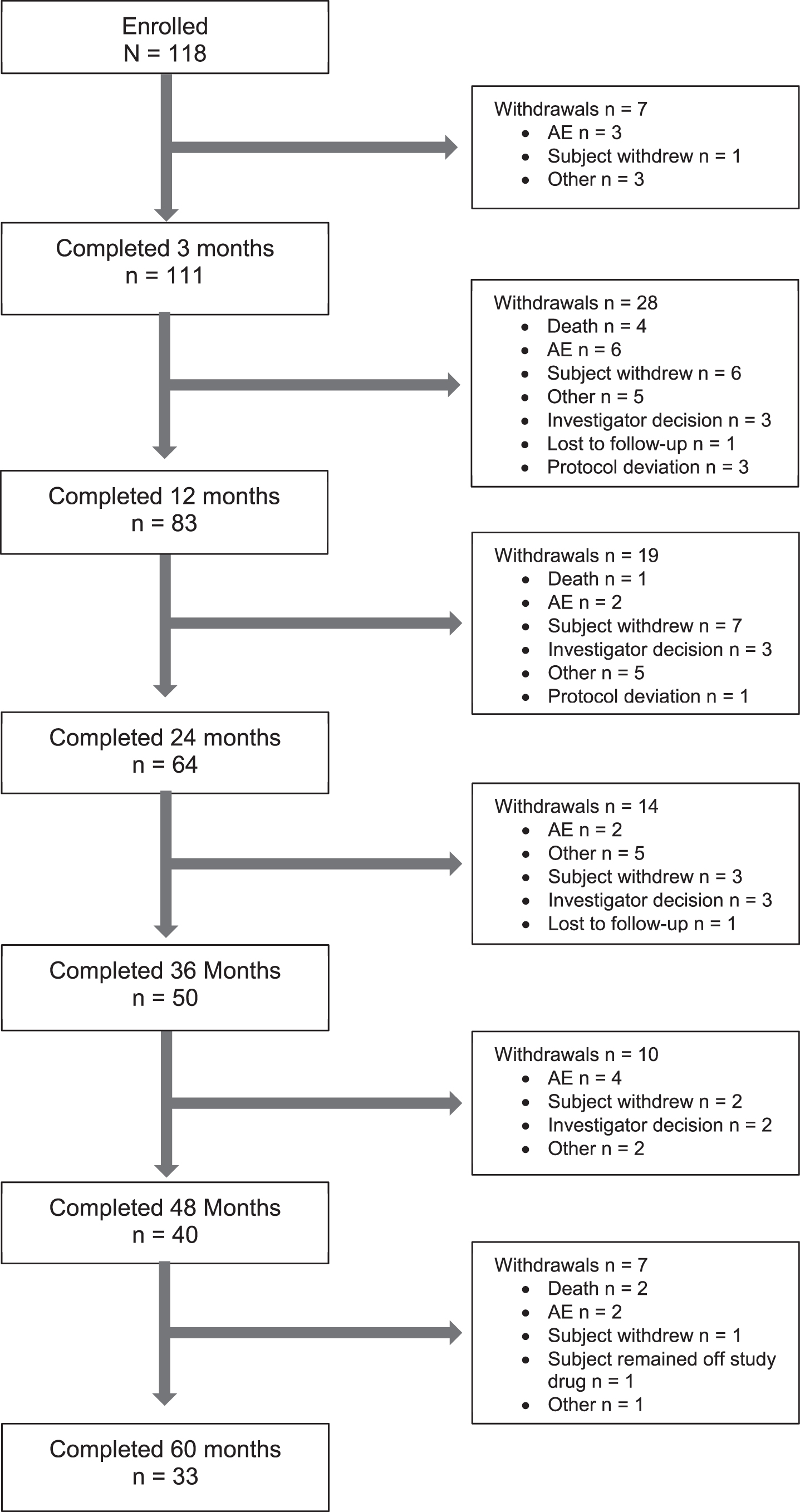
Participant disposition throughout study. Figure includes results for participants who were originally enrolled and successfully completed the double-blind HART Study and re-enrolled into the Open-HART Study.
Table 1 depicts baseline demographics and concomitant psychoactive medications taken during the study. The majority of psychoactive medications were antidepressants (notably sertraline and paroxetine, 26.3% and 16.1%), with anxiolytics also common (clonazepam and lorazepam, 22% and 12.7%, respectively). Sleep aids were frequently used (trazodone 10.2%; zolpidem 9.3%; mirtazapine 7.6%), as was the mood-stabilizing agent valproate (9.3%).
Baseline characteristics and concomitant medications
Table includes results for All patients (participants who were originally enrolled and successfully completed the double-blind HART Study and reenrolled into the Open-HART Study), 60 Months completers and early HD subgroup with baseline TFC 9–13; CAG not available for all participants. SD, standard deviation; TFC, total functional capacity.
Safety
Table 2 lists cumulative adverse events for the entire study (N = 118). 89.8% of participants reported at least one AE over the study interval, with the majority experiencing mild (16.1%) or moderate (44.9%) events. Adverse events reported by more than 10% of participants are displayed, with features of HD predominantly represented (falls, weight loss, anxiety, insomnia, depression, chorea) as well as trauma (lacerations, contusions). Thirty-four participants, or 28.8% of the total cohort, reported an event judged as severe.
Overview of adverse events
*Table includes results for participants who were originally enrolled and successfully completed the double-blind HART Study and reenrolled into the Open-HART Study. **Treatment-related determined by investigator. AE, adverse event; QTcF, Fridericia-corrected QT interval.
Twenty-eight patients (23.7%) reported at least one SAE. Of the 67 events of SAEs reported in Open-HART, 7 were considered at least possibly treatment-related by investigators: anxiety, multiple myeloma, retinal detachment, seizure, loss of consciousness, subdural hematoma, and delirium (Table 3). At 48, and 60 months, common AE classes were Injury (particularly falls) and Psychiatric Disorders (Table 4). Falls were reported by a higher percentage of participants at 48 and 60 months (25.0% and 30.3%, respectively) compared to earlier in the study (10.9% and 22.0% at 24 and 36 months, respectively). Chorea as an adverse event was not common at 48 and 60 months (5.0% and 3.0%). Reduced weight was more common earlier in the study (24-month period).
Serious adverse events
*Table includes results for participants who were originally enrolled and successfully completed the double-blind HART Study and reenrolled into the Open-HART Study. **Treatment-related determined by investigator. AE, adverse event.
Number of participants reporting adverse events by year
n value represents the number of patients completing the indicated year of the study. AE, adverse event.
Over 60 months, 16 participants experienced QTcF prolongation above 450 milliseconds (13.6%), with 3 of these occurring since the 36-month analysis; 5 participants had an absolute change of 60 milliseconds or greater above baseline (4.2%), with 5 after the 36-month analysis. These observations were not associated with any cardiac adverse events or related symptoms. No patient had a QTcF interval duration greater than 500 msec at any time point. A single AE of arrhythmia was reported by 60 months, and was the only instance in the dataset. No syncopal AEs were reported after 36 months, and no MI events beyond 12 months. One seizure was reported in both the 48 and 60-month intervals; these were classified as unrelated to study drug. Mean QTcF values and heart rate remained stable over the 60-month period.
Seven deaths occurred in the 60-month period, with two following the 3-year visit. Of these, one participant expired of multiple myeloma at 4.3 years while the other was a completed suicide after 4.2 years in the study. None of the adverse events resulting in death were considered by the investigator to be related to the study drug.
Exploratory efficacy
Exploratory efficacy measures for TFC and TMS data are indicated in Table 5. The rates of decline in TFC (lower scores indicate worsening) and TMS (higher scores indicate worsening) were evaluated for the entire cohort (N = 118), for the 60 months completers (N = 33), and for the early HD subgroup (N = 55, baseline TFC 9–13) in Open-HART. For comparison, an early HD cohort (TFC 9–13) from 2CARE participants receiving placebo was also analyzed for TFC change over 60 months.
Exploratory efficacy analyses for UHDRS TFC and TMS
Table includes results for all patients (participants who were originally enrolled and successfully completed the double-blind HART Study and reenrolled into the Open-HART Study), 60 Months completers and early HD subgroup with baseline TFC 9–13. p-values shown compare the mean change from baseline between the Open-HART and 2CARE studies at each time-point. n value represents the number of patients who completed the indicated year of the study. SE, standard error; UHDRS, Unified Huntington’s Disease Rating Scale.
Demographic characteristics (age, sex, weight) for early HD participants in Open-HART and 2CARE at baseline, 48 and 60 months were similar (Supplementary Table 1). The baseline mean (SE) TFC was 8.4 (0.2) for the entire cohort, 8.8 (0.5) for the 60 months completers, and 10.6 (0.2) for the early HD subgroup (TFC 9–13). Mean baseline TFC (SE) was 11.1 (0.1) for the 2CARE placebo group (n = 303, early HD TFC 9–13). Comparing the early HD subgroups (baseline TFC 9–13) between Open-HART patients receiving pridopidine 45 mg BID and patients from 2CARE receiving placebo, mean change from baseline in TFC at 24, 36, 48 and 60 months was –1.2 (0.4), –2.1 (0.4), –2.0 (0.6) and –1.8 (0.5) for Open HART and –2.0 (0.1), –2.7 (0.2), –3.8 (0.2) and –5.0 (0.3) for 2CARE.
Open-HART had nominally significantly slower TFC decline than 2CARE at 48 months (–2.0 (0.6) vs. –3.8 (0.2), p = 0.01) and 60 months (–1.8 (0.5) vs. –5.0 (0.3), p = 0.001) (Table 5). In Open-HART, TFC remained stable (no worsening) between 48 and 60 months, with a mean (SE) of 6.8 (0.6) for the entire cohort and 8.9 (0.5) for the early HD subgroup. Over this same interval, the 2CARE early cohort was not stable, deteriorating by –1.2 points. To address the influence of dropouts on TFC over the course of the study, we conducted a sensitivity analysis using MMRM and multiple imputation for earlier-stage participants (TFC 9–13) from both Open-HART and 2CARE (Table 6). With this approach, observations for the baseline TFC 9–13 group achieving nominal significance in the non-imputed analysis remained nominally significant, again showing less TFC decline for Open HART participants at 48 and 60 months compared to 2CARE (p = 0.01, 0.001, respectively). MMRM analysis indicated minimal deterioration in TFC between 36 and 60 months, with mean TFC (SE) of 8.5 (0.4), 8.4 (0.5) and 8.2 (0.5) at 36, 48 and 60 months, respectively.
Exploratory efficacy analyses for UHDRS TFC and TMS with multiply imputed data
p-values shown compare the mean change from baseline between the Open-HART and 2CARE studies at each time-point. SE, standard error; UHDRS, Unified Huntington’s Disease Rating Scale.
Less decline was seen in Open HART participants at 60 months in each of the TFC domains; when corrected for multiple comparisons, ADLs, finances, care level, and domestic chores were significantly different than 2CARE early TFC cohorts (–0.3 vs. –1.0; –0.4 vs –1.3; –0.1 vs. –0.5; and –0.4 vs –1.0, respectively, all p < 0.01) (Table 7). Sensitivity analyses for baseline TFC 9–13 participants from both Open HART and 2CARE resulted in preservation of these same nominally significant findings (Table 8).
Mean change in total functional capacity domains for early HD cohorts (TFC 9–13) Completing 60 months from open-HART and 2CARE
Table includes results for early HD participants from the Open-HART study and 2CARE trial with baseline TFC 9–13. p-values shown compare the change from baseline between the Open-HART and 2CARE studies at each time point.
Exploratory efficacy analyses for UHDRS TFC domains with multiply imputed data
Table includes results for early HD participants from the Open-HART study and 2CARE trial with baseline TFC 9–13. * Indicates the p-value comparing the change from baseline between the Open-HART and 2CARE studies at each time-point.
In Open-HART, mean (SE) baseline TMS was 38.7 (1.5) for entire cohort, 32.4 (2.4) for the 60 months completers, and 32.9 (1.7) for the early HD cohort. The 60-month completers and early HD cohorts had lower mean baseline TMS scores compared to the entire cohort. The early-stage HD cohort in Open-HART had a higher (more impaired) mean (SE) TMS score at baseline compared to the early stage HD 2CARE cohort (32.9 (1.7) Open-HART vs 27.4 (0.8) 2CARE). In both the Open-HART and the 2CARE trials, TMS increased over 5 years, indicating motor deterioration. The mean TMS change for the early HD Open-HART cohort was consistently smaller compared to early HD in the 2CARE placebo arm. Early HD in Open-HART showed less TMS deterioration than 2CARE at 24 months (2.9 (1.7) vs 7.4 (0.7) p = 0.05), 48 months (9.1(2.5) vs 14.3(1.1), p = 0.05) and 60 months (10.5(2.6) vs 18.5(1.5), p = 0.06). TMS scores remained stable in the early HD Open-HART cohort at months 36, 48 and 60 (mean TMS 36.5(2.6), 35.9(3.3) and 36.9(3.7) respectively), though the available sample was decreasing progressively with time. Over this same interval the 2CARE early cohort appeared to worsen, with TMS mean (SE) of 36.9 (1.3), 41.5 (1.5) and 46.0 (2.0) at 36, 48 and 60 months. To analyze longitudinal TMS change in the baseline TFC 9–13 group while accounting for dropouts, we again conducted a MMRM sensitivity analysis with multiple imputation (Table 6). The mean TMS decline from baseline was again consistently smaller in Open-HART compared to 2CARE early HD participants using the MMRM analysis. Early HD in Open-HART showed less TMS decline than 2CARE at 24 months (3.8 (1.7) vs 7.3 (0.7)), 36 months (8.3(1.8) vs 10.1(0.7)), 48 months (10.6 (2.2) vs 15.0 (0.9)) and 60 months (14.2(2.5) vs 19.0 (1.0)). Sensitivity analyses for differences from baseline between Open-HART and 2CARE for the early cohort at 48 and 60 months again trended towards nominally significant values (p = 0.07 at 48 months and p = 0.08 at 60 months).
Additional exploratory analyses were conducted on “early initiators” (patients previously exposed to pridopidine in HART) and “late initiators” (patients starting pridopidine for the first time in Open-HART) as shown in Supplementary Table 2. The early cohort was substantially larger than the late cohort (n = 87, vs. n = 31), while the late cohort had relatively more female participants (64.5%, vs 48.3%). Psychoactive medications, adverse events and the proportion of subjects reporting serious adverse events were generally similar between groups (Supplementary Tables 3–5). Early initiators showed less decline at Months 48 and 60 in TFC compared to the late initiators, though TMS scores were variable; small numbers in the late initiator group over time prevent meaningful comparisons (Supplementary Table 6). By 48 months, mean (SE) TFC change from baseline was –1.7 (0.4) for the early initiators (n = 31) compared to –2.8 (0.9) in the late initiators group (n = 9). Between Month 48 and 60, there was an apparent improvement in TFC in the late initiators group [–2.8 (0.9) decline at 48 months vs –2.0 (1.0) decline at 60 months], most likely due to the discontinuation of 3 out of 9 patients (33%). As a result, at 60 months, the change from baseline in TFC was comparable between early initiators [n = 26, –1.9 (0.4)] and late initiators [n = 6, –2.0 (1.0)].
DISCUSSION
In this open label pridopidine extension study, representing the longest treatment duration to date, pridopidine remained safe and tolerable over 60 months. No unexpected safety or tolerability issues emerged in the period after the previous 36-month report [13]. As in the prior analysis, no apparent cumulative toxicity appeared to emerge over time, and laboratory, ECG, and vital signs remained stable over the study period. QTcF did not appear to worsen over time with concomitant use of QT-prolonging medications (antidepressants). The majority of adverse events noted by more than 10% of subjects were related to HD phenotype or likely secondary to trauma, an expected feature of HD. Suicidality did not emerge as a concern, as the observed instance of completed suicide is not unexpected in HD. In the period from 36 months onward, falls continued to be the most commonly occurring AE, consistent with progressive gait instability. Overall, the 5-year Open-HART data suggests that 45 mg BID (90 mg/daily) pridopidine has a benign safety and tolerability profile in HD patients, and is comparable with the safety profile reported in prior HD trials with pridopidine (HART, MermaiHD and PRIDE-HD) [12, 18].
While TFC declined over time in Open-HART exploratory analyses, mean annual TFC decline over 60 months was lower in pridopidine-treated participants (0.4 points/year) compared to the placebo group from 2CARE, where an expected average decline of 1 point per year was observed. TFC scores in Open-HART appeared to remain stable for all participants between 48 and 60 months at 6.8 (0.6) points, while also remaining stable for early HD participants (TFC 9–13) between 36, 48, and 60 months [mean (SE) of 8.8 (2.3), 8.9 (0.5), 8.9 (0.5), respectively]. This observation of stability in early HD between 36, 48 and 60 months remained after sensitivity analysis to account for dropouts [8.5 (0.4), 8.5 (0.5), and 8.2 (0.5), respectively] and was nominally significantly different from 2CARE at 48 months [(8.4 (0.5) Open-HART vs. 7.4 (0.2) 2CARE] and 60 months [(8.2 (0.5) Open-HART vs. 6.5 (0.2) 2CARE]. TFC stability in Open-HART compared to 2CARE at 48 and 60 months for the baseline TFC 9–13 group in both non-imputed and imputed analyses suggest this finding may be robust despite the progressive dropout of participants over the study period. TMS at 48 and 60 months also appeared to remain stable, averaging approximately 45 points. For early HD patients (TFC 9–13) from Open-HART, TMS appeared stable at 36, 48 and 60 months (mean (SE) 36.5(2.6), 35.9(3.3) and 36.9 (3.7), respectively). In 2CARE, TMS in the TFC 9–13 group continued to deteriorate, with respective mean (SE) of 36.9 (1.3), 41.5 (1.5) and 46.0 (2.0) at 36, 48 and 60 months. With sensitivity analysis to account for missing data, the trend toward nominally significant difference in TMS from baseline at 48 and 60 months remained similar to non-imputed data, although comparative sample sizes across the studies were still substantially different (N = 303 2CARE, N = 55 Open-HART).
The period from 48 to 60 months appears to demonstrate stable TFC and TMS scores, although small numbers and the open-label nature of the study make it inconclusive as to how much of this exploratory finding reflects drug effect on functional status or motor ability, placebo effect, or randomness. It is possible that the progressively decreasing cohort size over time produced a confounding effect, with participants having less aggressive disease over-represented in later analyses. The Open-HART subgroup able to complete 60 months had comparable baseline demographic characteristics (age, weight, sex) to the entire Open-HART cohort but mildly better TFC and TMS scores at study entry [TFC 8.8 (0.5), 60-month completers vs 8.4 (0.2), total; TMS 27.4 (0.8), 60-month completers vs. 32.4 (2.4), total), making it plausible that greater initial functional reserve or less motor disability promoted better longevity in the study. It is conceivable that trial participation had a behaviorally reinforcing or stabilizing effect over time, but this remains conjecture and outside the scope of our analysis. The relatively static TFC performance at Months 48 and 60 may be consistent with a stabilizing or protective effect of pridopidine, which is intriguing to consider given emerging preclinical evidence of neuroprotection via the S1R. This consideration remains hypothetical given the limitations of these data.
Overall, 45 mg BID pridopidine continued to be safe and tolerable during long-term use in the HD population. These Open-HART data suggesting less TFC decline over time in early-stage participants compared to matched 2CARE placebo historical controls are consistent with observations from the recently completed randomized, double-blind, placebo-controlled PRIDE-HD trial, where pridopidine 45 mg BID was associated with less TFC decline at 52 weeks compared to placebo [18]. Emerging data regarding potentially neuroprotective effects of pridopidine mediated by the S1R raise the possibility that some component of long-term TFC performance in Open-HART is driven by an underlying biological effect. This hypothesis, with converging trial data and a plausible mechanism substantiated by preclinical findings, warrants further testing of the 45 mg BID dosage in early-disease cohorts (i.e., TFC 9-13) in adequately powered, randomized, double-blind studies.
CONFLICT OF INTEREST
AM, KK, and PA have previously received grant support from Teva. MG, VA, IG, SP, and MH are previous employees of Teva. MFG, SG, and JH are employees of Teva.